Synthesis and Functionalization of a 1,2-Bis(trimethylsilyl)-1,2-disilacyclohexene That Can Serve as a Unit of cis-1,2-Dialkyldisilene


Abstract
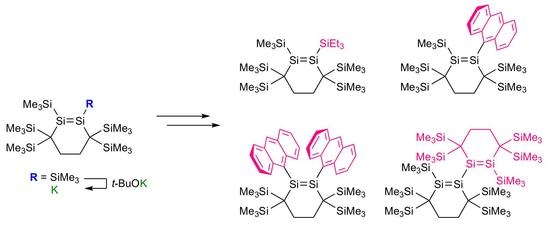
:
1. Introduction
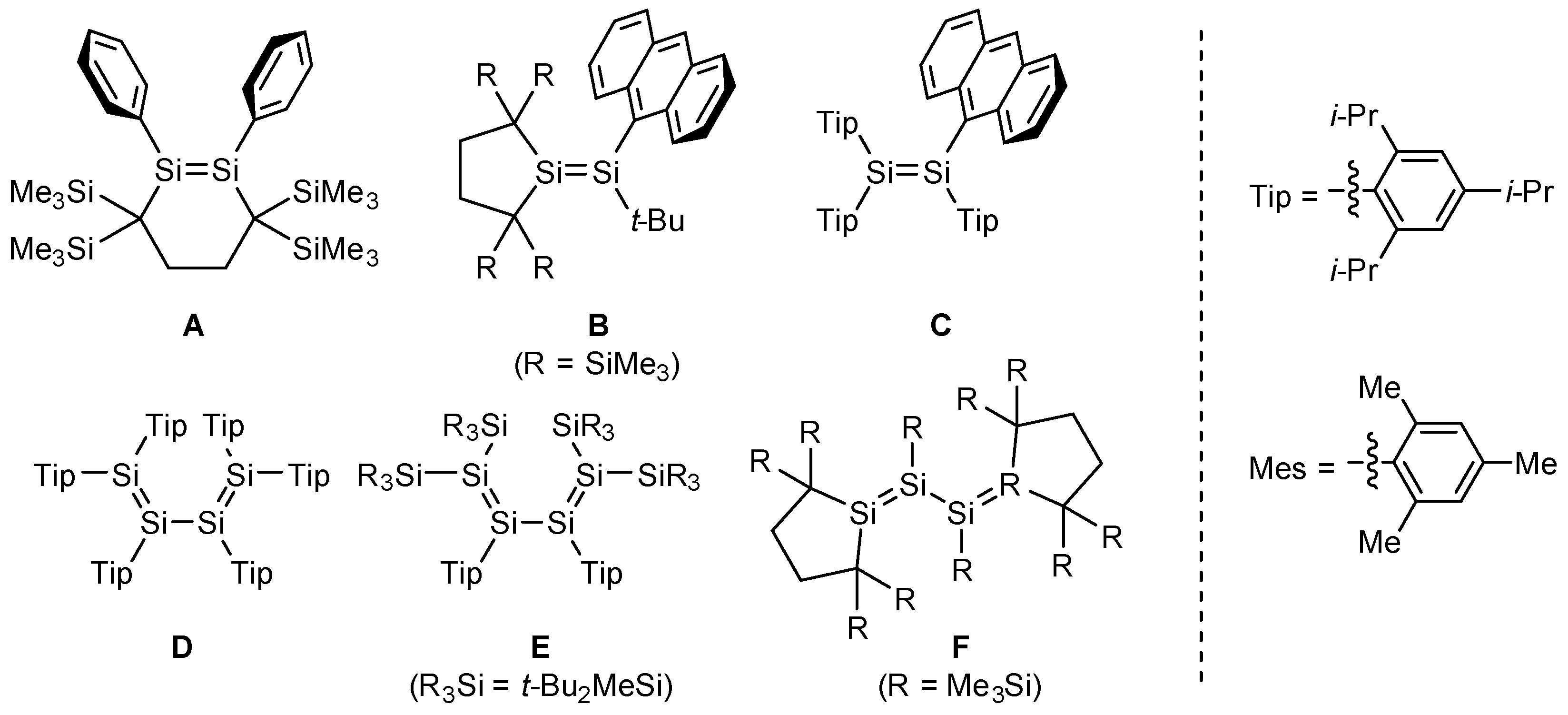
2. Results and Discussion
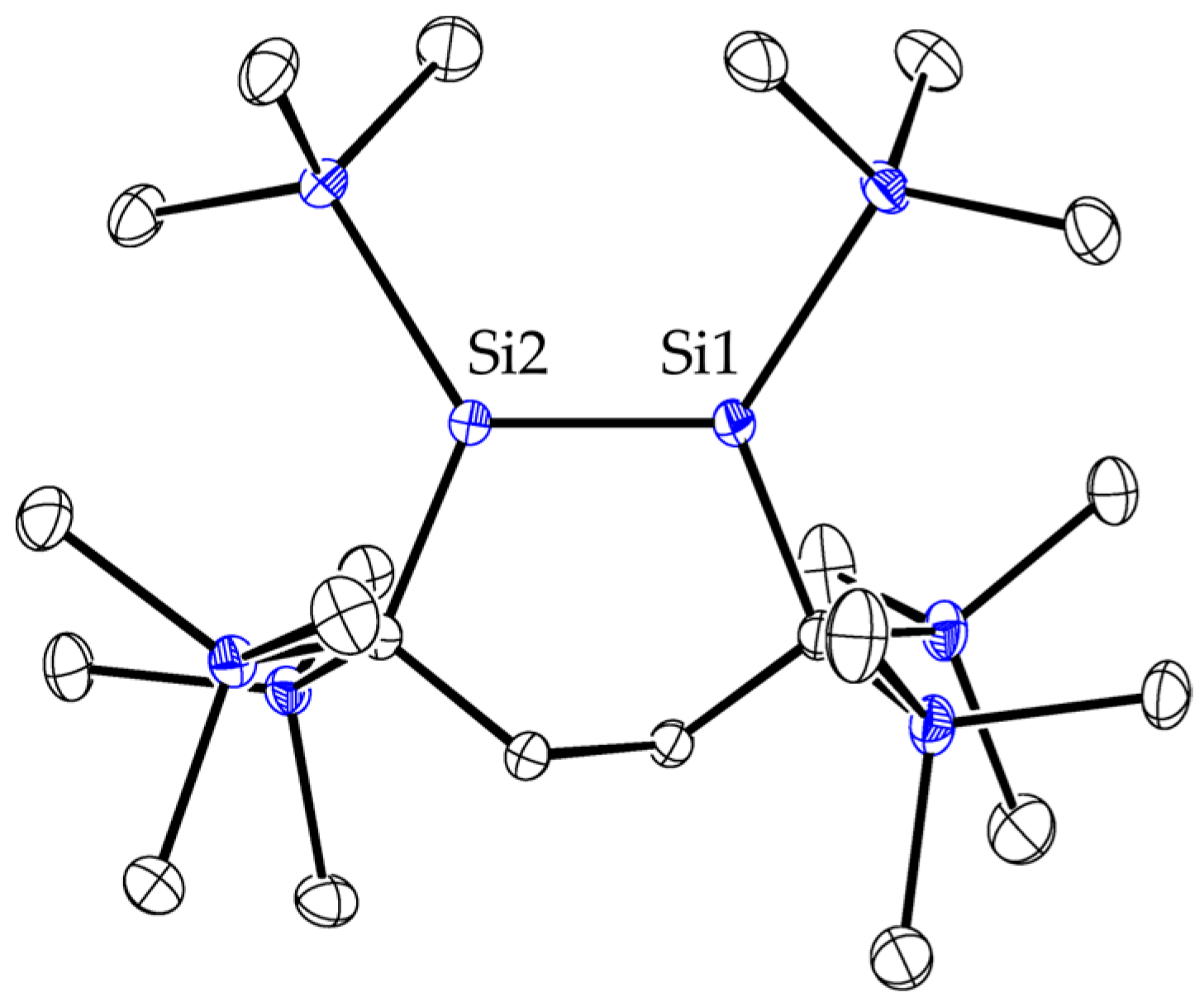
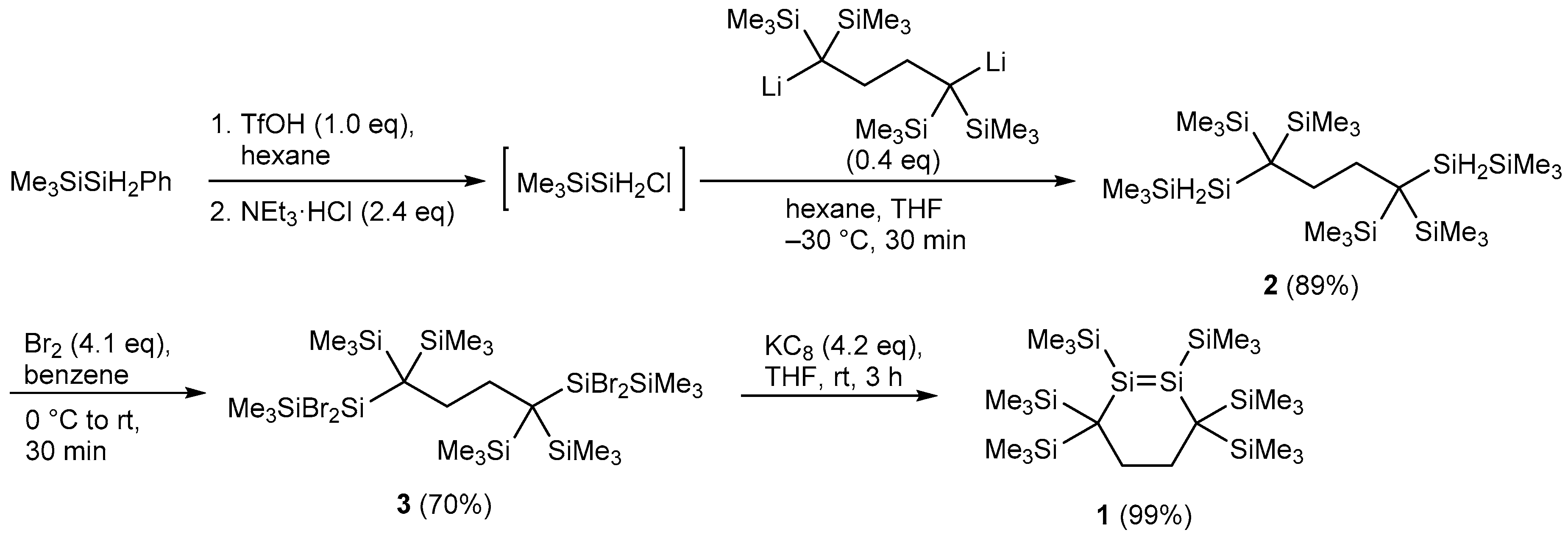
2.1. Synthesis and Molecular Structure of Disilacyclohexene 1
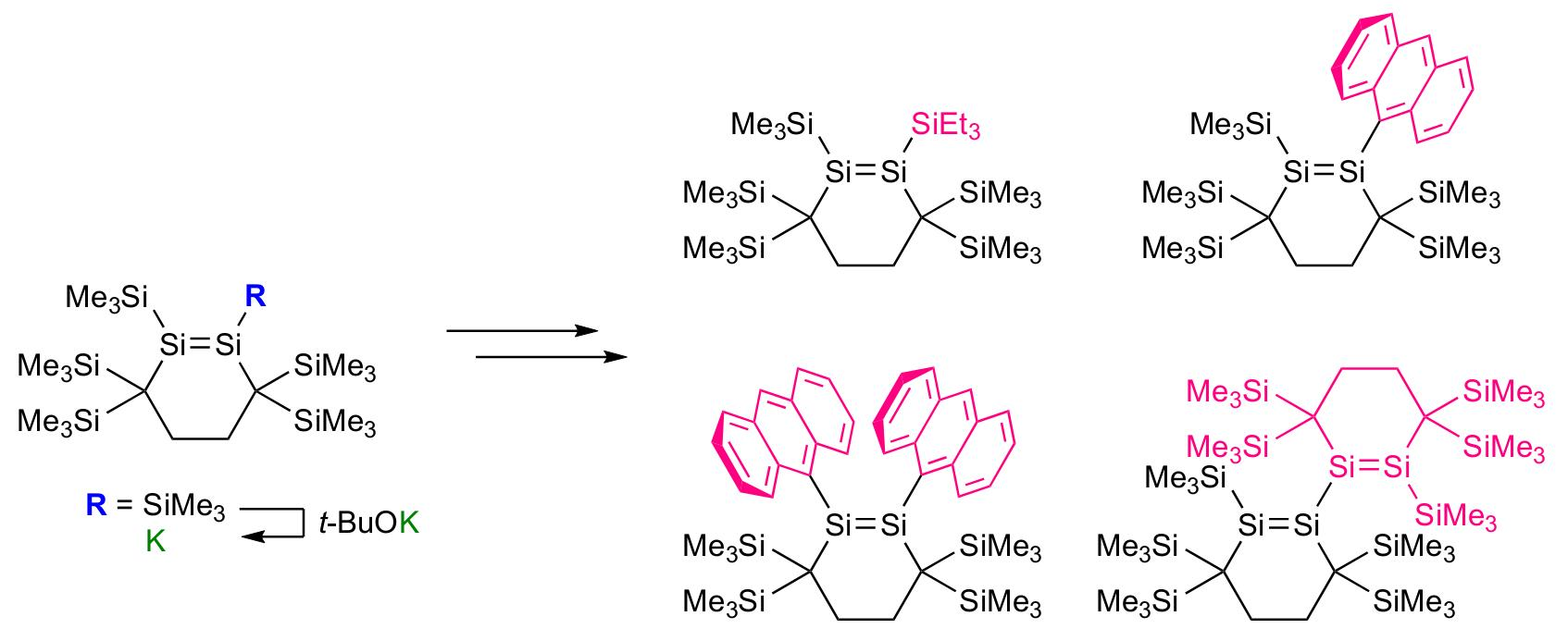
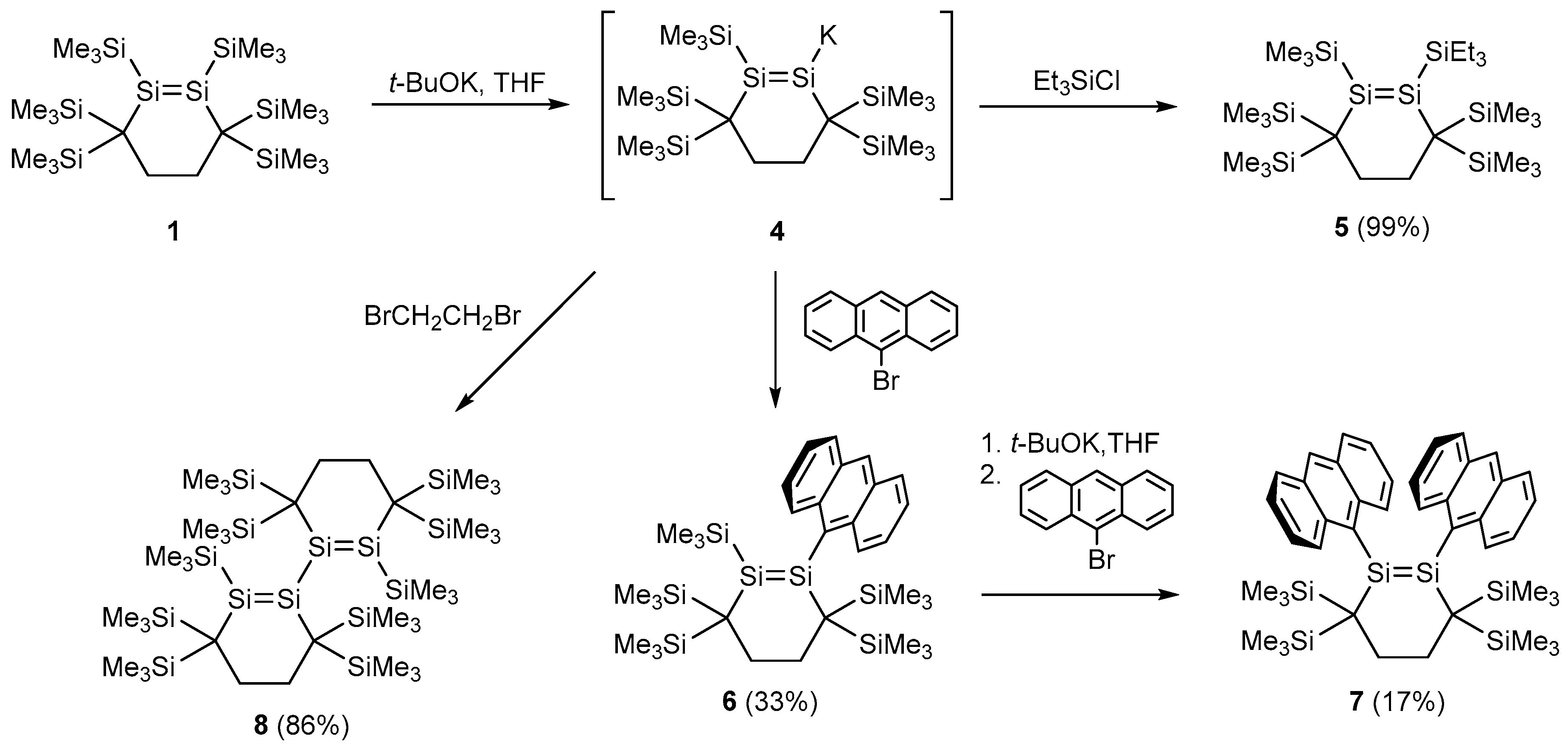
2.2. Conversion of 1 to Disilenide 4 and Functionalized Disilenes
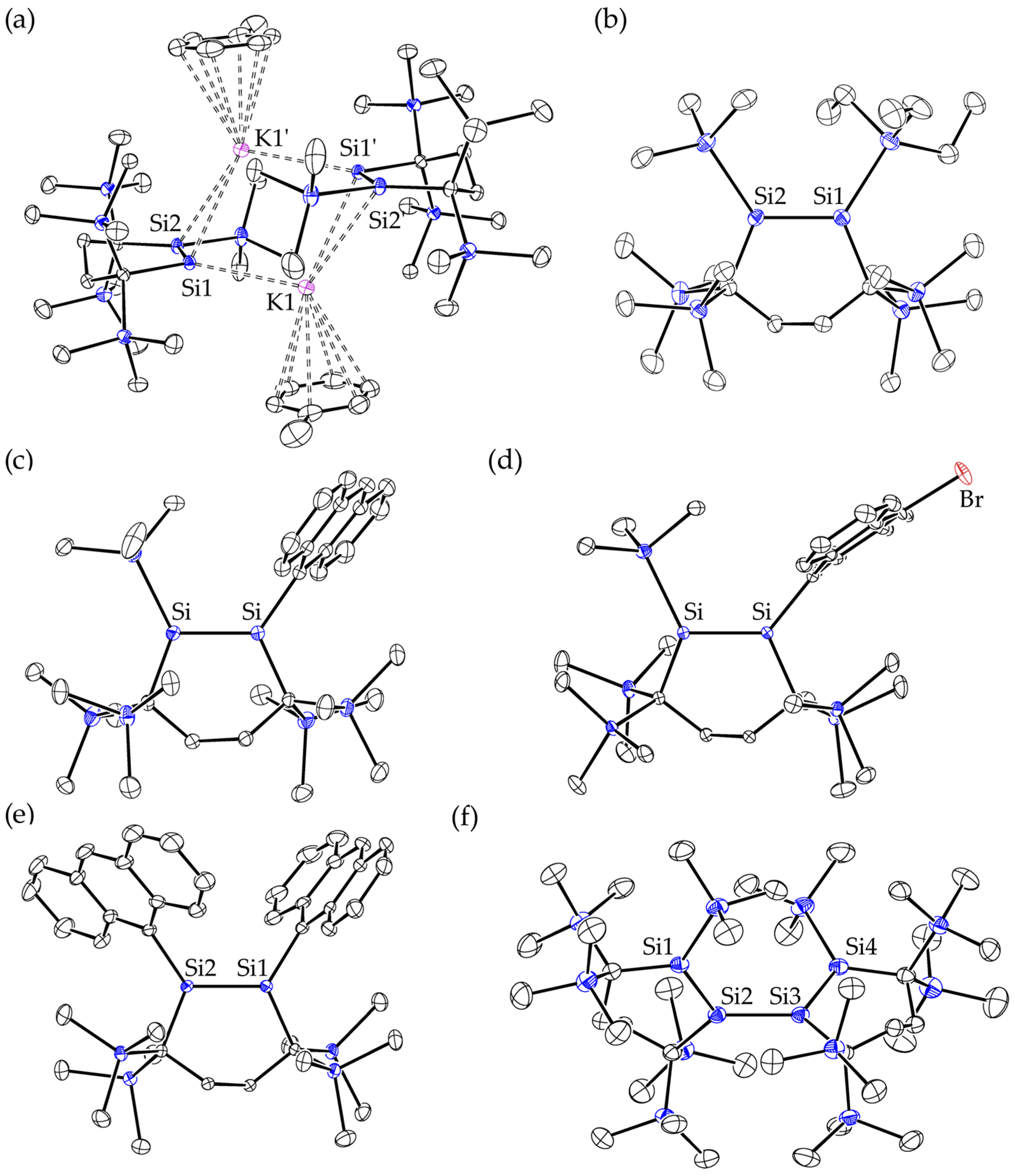
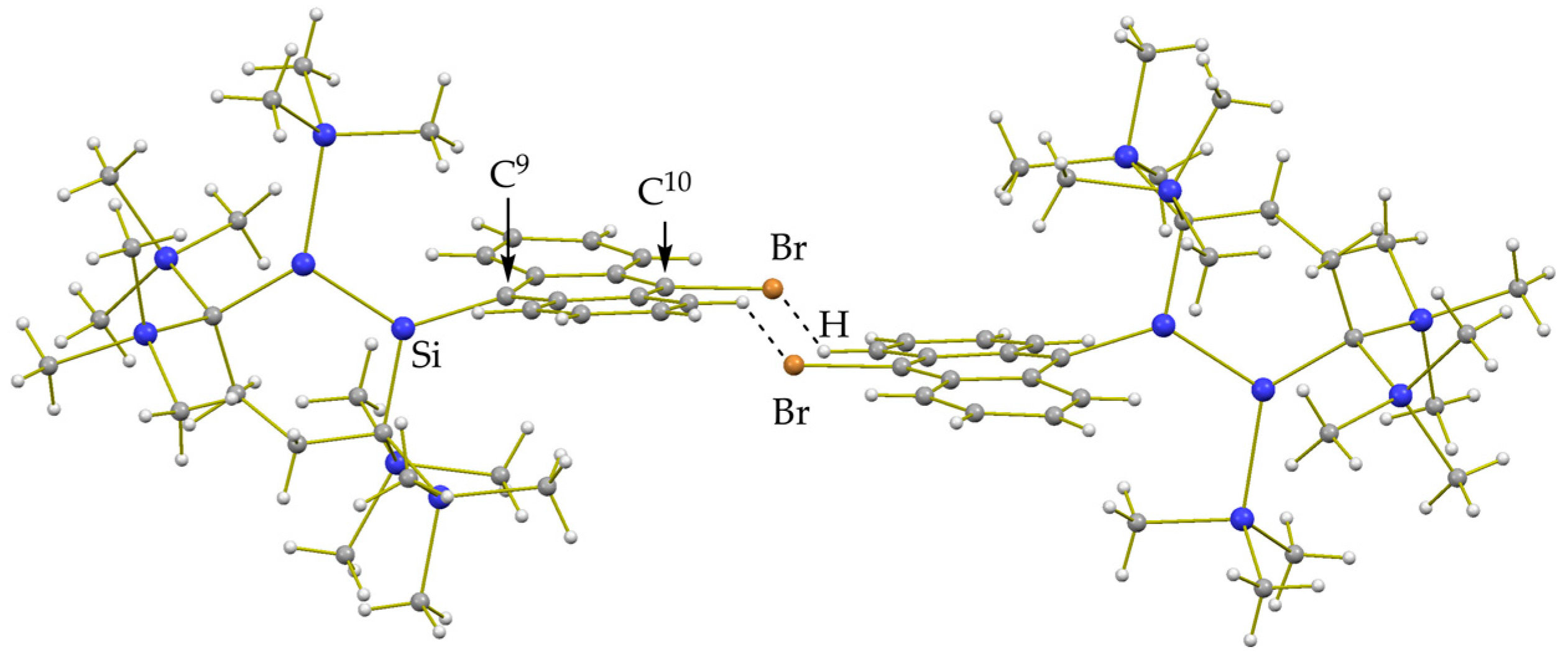
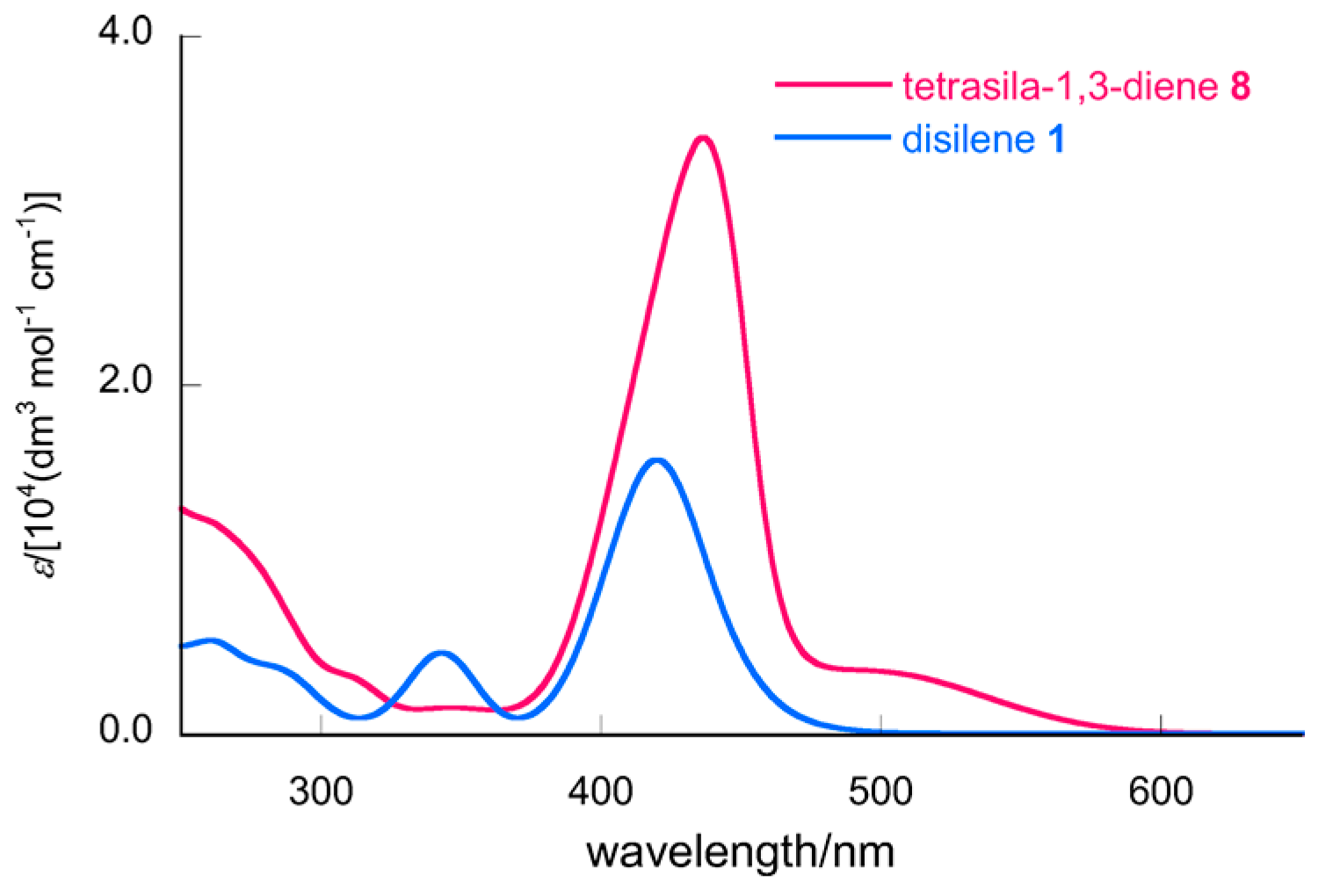
2.3. Molecular Structures of Disilenes 5–8
3. Materials and Methods
3.1. General Procedure
3.2. Materials
3.3. Preparation of 1,4-Bis(2,2,2-trimethyldisilanyl)-1,1,4,4-tetrakis(trimethylsilyl)butane 2
3.4. Preparation of 1,4-Bis(1,1-dibromo-2,2,2-trimethyldisilanyl)-1,1,4,4-tetrakis(trimethylsilyl)butane 3
3.5. Synthesis of 1,2,3,3,6,6-Hexakis(trimethylsilyl)-1,2-disilacyclohexene 1
3.6. Synthesis of Potassium 2,3,3,6,6-Pentakis(trimethylsilyl)-1,2-disilacyclohexen-1-ide 4
3.7. Synthesis of 1-Triethylsilyl-2,3,3,6,6-pentakis(trimethylsilyl)-1,2-disilacyclohexene 5
3.8. Synthesis of 1-(9-Anthryl)-2,3,3,6,6-pentakis(trimethylsilyl)-1,2-disilacyclohexene 6
3.9. Synthesis of 1-(10-Bromo-9-anthryl)-2,3,3,6,6-pentakis(trimethylsilyl)-1,2-disilacyclohexene 6Br
3.10. Synthesis of 1,2-Di(9-anthryl)-3,3,6,6-tetrakis(trimethylsilyl)-1,2-disilacyclohexene 7
3.11. Synthesis of 1,4-Bis(trimethylsilyl)tetrasila-1,3-diene 8
3.12. Single Crystal X-ray Diffraction Analyses
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Okazaki, R.; West, R. Chemistry of stable disilenes. Adv. Organomet. Chem. 1996, 39, 231–273. [Google Scholar]
- Kira, M.; Iwamoto, T. Progress in the chemistry of stable disilenes. Adv. Organomet. Chem. 2006, 54, 73–148. [Google Scholar]
- Lee, V.Y.; Sekiguchi, A. Heavy analogs of alkenes, 1,3-dienes, allenes and alkynes: Multiply bonded derivatives of Si, Ge, Sn and Pb. In Organometallic Compounds of Low-Coordinate Si, Ge, Sn and Pb: From Phantom Species to Stable Compounds; Wiley-VCH: Chichester, Germany, 2010; pp. 199–334. [Google Scholar]
- Scheschkewitz, D. Anionic reagents with silicon-containing double bonds. Chem. Eur. J. 2009, 15, 2476–2485. [Google Scholar] [CrossRef] [PubMed]
- Abersfelder, K.; Scheschkewitz, D. Synthesis of homo- and heterocyclic silanes via intermediates with Si=Si bonds. Pure Appl. Chem. 2010, 82, 595–602. [Google Scholar] [CrossRef]
- Scheschkewitz, D. The versatile chemistry of disilenides: Disila analogues of vinyl anions as synthons in low-valent silicon chemistry. Chem. Lett. 2011, 40, 2–11. [Google Scholar] [CrossRef]
- Präsang, C.; Scheschkewitz, D. Silyl anions. Struct. Bonding 2013, 156, 1–47. [Google Scholar]
- Präsang, C.; Scheschkewitz, D. Reactivity in the periphery of functionalised multiple bonds of heavier group 14 elements. Chem. Soc. Rev. 2016, 45, 900–921. [Google Scholar] [CrossRef] [PubMed]
- Rammo, A.; Scheschkewitz, D. Functional disilenes in synthesis. Chem. Eur. J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T.; Kobayashi, M.; Uchiyama, K.; Sasaki, S.; Nagendran, S.; Isobe, H.; Kira, M. Anthryl-substituted trialkyldisilene showing distinct intramolecular charge-transfer transition. J. Am. Chem. Soc. 2009, 131, 3156–3157. [Google Scholar] [CrossRef] [PubMed]
- Scheschkewitz, D. A silicon analogue of vinyllithium: Structural characterization of a disilenide. Angew. Chem. Int. Ed. 2004, 43, 2965–2967. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, M.; Sanuki, K.; Inoue, S.; Sekiguchi, A. Disilenyllithium from tetrasila-1,3-butadiene: A silicon analogue of a vinyllithium. Organometallics 2004, 23, 3088–3090. [Google Scholar] [CrossRef]
- Ichinohe, M.; Sanuki, K.; Inoue, S.; Sekiguchi, A. Tetrasila-1,3-butadiene and its transformation to disilenyl anions. Silicon Chem. 2006, 3, 111–116. [Google Scholar] [CrossRef]
- Meltzer, A.; Majumdar, M.; White, A.J.P.; Huch, V.; Scheschkewitz, D. Potential protecting group strategy for disila analogues of vinyllithiums: Synthesis and reactivity of a 2,4,6-trimethoxyphenyl-substituted disilene. Organometallics 2013, 32, 6844–6850. [Google Scholar] [CrossRef]
- Abersfelder, K.; Zhao, H.; White, A.J.P.; Präsang, C.; Scheschkewitz, D. Synthesis of the first homoleptic trisilaallyl chloride: 3-Chloro-1,1,2,3,3-pentakis(2′,4′,6′-triisopropylphenyl)trisil-1-ene. Z. Anorg. Allg. Chem. 2015, 641, 2051–2055. [Google Scholar] [CrossRef]
- Akasaka, N.; Fujieda, K.; Garoni, E.; Kamada, K.; Matsui, H.; Nakano, M.; Iwamoto, T. Synthesis and functionalization of a 1,4-bis(trimethylsilyl)tetrasila-1,3-diene through the selective cleavage of Si(sp2)-Si(sp3) bonds under mild reaction conditions. Organometallics 2018, 37, 172–175. [Google Scholar] [CrossRef]
- Marschner, C. Silicon-centered anions. In Organosilicon Compounds Theory and Experiment (Synthesis); Lee, V.Y., Ed.; Academic Press: Oxford, UK, 2017; pp. 295–360. [Google Scholar]
- Abe, T.; Iwamoto, T.; Kira, M. A stable 1,2-disilacyclohexene and its 14-electron palladium(0) complex. J. Am. Chem. Soc. 2010, 132, 5008–5009. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Allen, L.C. Group IV double bonds: Shape deformation and substituent effects. J. Am. Chem. Soc. 1990, 112, 1039–1041. [Google Scholar] [CrossRef]
- Karni, M.; Apeloig, Y. Substituent effects on the geometries and energies of the Si=Si double bond. J. Am. Chem. Soc. 1990, 112, 8589–8590. [Google Scholar] [CrossRef]
- Apeloig, Y.; Müller, T. Do silylenes always dimerize to disilenes? Novel silylene dimers with unusual structures. J. Am. Chem. Soc. 1995, 117, 5363–5364. [Google Scholar] [CrossRef]
- Goldberg, D.E.; Hitchcock, P.B.; Lappert, M.F.; Thomas, K.M.; Thorne, A.J.; Fjeldberg, T.; Haaland, A.; Schilling, B.E.R. Subvalent Group 4B metal alkyls and amides. Part 9. Germanium and tin alkene analogues, the dimetallenes M2R4[M = Ge or Sn, R = CH(SiMe3)2]: X-ray structures, molecular orbital calculations for M2H4, and trends in the series M2R′4[M = C, Si, Ge, or Sn; R′ = R, Ph, C6H2Me3-2,4,6, or C6H3Et2-2,6]. J. Chem. Soc. Dalton Trans. 1986, 2387–2394. [Google Scholar]
- West, R.; Cavalieri, J.D.; Buffy, J.J.; Fry, C.; Zilm, K.W.; Duchamp, J.C.; Kira, M.; Iwamoto, T.; Müller, T.; Apeloig, Y. A solid-state NMR and theoretical study of the chemical bonding in disilenes. J. Am. Chem. Soc. 1997, 119, 4972–4976. [Google Scholar] [CrossRef]
- Obeid, N.M.; Klemmer, L.; Maus, D.; Zimmer, M.; Jeck, J.; Bejan, I.; White, A.J.P.; Huch, V.; Jung, G.; Scheschkewitz, D. (Oligo)Aromatic Species With One or Two Conjugated Si=Si Bonds: Near-IR Emission of Anthracenyl-Bridged Tetrasiladiene. Dalton Trans. 2017, 46, 8839–8848. [Google Scholar] [CrossRef] [PubMed]
- Weidenbruch, M.; Willms, S.; Saak, W.; Henkel, G. Hexaaryltetrasilabuta-1,3-diene: A molecule with conjugated Si=Si double bonds. Angew. Chem. Int. Ed. Engl. 1997, 36, 2503–2504. [Google Scholar] [CrossRef]
- Uchiyama, K.; Nagendran, S.; Ishida, S.; Iwamoto, T.; Kira, M. Thermal and photochemical cleavage of Si=Si double bond in tetrasila-1,3-diene. J. Am. Chem. Soc. 2007, 129, 10638–10639. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T.; Ishida, S. Multiple bonds with silicon: Recent advances in synthesis, structure, and functions of stable disilenes. In Structure and Bonding: Functional Molecular Silicon Compounds II; Scheschkewitz, D., Ed.; Springer: Cham, Switzerland, 2017; Volume 156, pp. 125–202. [Google Scholar]
- Kosai, T.; Iwamoto, T. Stable push–pull disilene: Substantial donor–acceptor interactions through the Si=Si double bond. J. Am. Chem. Soc. 2017, 139, 18146–18149. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.J.; Abersfelder, K.; White, A.J.; Majumdar, M.; Scheschkewitz, D. Transmetallation reactions of a lithium disilenide. Chem. Commun. 2012, 48, 6595–6597. [Google Scholar] [CrossRef] [PubMed]
- Auer, D.; Strohmann, C.; Arbuznikov, A.V.; Kaupp, M. Understanding substituent effects on 29Si chemical shifts and bonding in disilenes. A quantum chemical analysis. Organometallics 2003, 22, 2442–2449. [Google Scholar] [CrossRef]
- Mazik, M.; Buthe, A.C.; Jones, P.G. C–H···Br, C–Br···Br, and C–Br···π interactions in the crystal structures of mesitylene- and dimesitylmethane-derived compounds bearing bromomethyl units. Tetrahedron 2010, 66, 385–389. [Google Scholar] [CrossRef]
- Safin, D.A.; Babashkina, M.G.; Robeyns, K.; Garcia, Y. C–H···Br–C vs. C–Br···Br–C vs. C–Br···N bonding in molecular self-assembly of pyridine-containing dyes. RSC Adv. 2016, 6, 53669–53678. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pati, N.N.; Kumar, B.S.; Chandra, B.; Panda, P.K. Unsymmetrical bipyrrole-derived β-tetraalkylporphycenes and C–H···Br–C interaction induced 2d arrays of the 2:1 supramolecular sandwich complex of their cis-/trans-dibromo isomers. Eur. J. Org. Chem. 2017, 741–745. [Google Scholar] [CrossRef]
- Ichinohe, M.; Arai, Y.; Sekiguchi, A.; Takagi, N.; Nagase, S. A new approach to the synthesis of unsymmetrical disilenes and germasilene: Unusual 29Si NMR chemical shifts and regiospecific methanol addition. Organometallics 2001, 20, 4141–4143. [Google Scholar] [CrossRef]
- Kosai, T.; Ishida, S.; Iwamoto, T. Heteroaryldisilenes: heteroaryl groups serve as electron acceptors for Si=Si double bonds in intramolecular charge transfer transitions. Dalton Trans. 2017, 46, 11271–11281. [Google Scholar] [CrossRef] [PubMed]
- Weitz, I.S.; Rabinovitz, M. The application of C8K for organic synthesis: Reduction of substituted naphthalenes. J. Chem. Soc. Perkin Trans. 1993, 1, 117–120. [Google Scholar] [CrossRef]
- Hengge, E.; Bauer, G.; Marketz, H. Darstellung und Eigenschaften einiger asymmetrisch substituierter 1,1,1-Trimethyldisilan-Verbindungen. Z. Anorg. Allg. Chem. 1972, 394, 93–100. [Google Scholar] [CrossRef]
- Kira, M.; Hino, T.; Kubota, Y.; Matsuyama, N.; Sakurai, H. Preparation and reactions of 1,1,4,4-tetrakis(trimethylsilyl)butane-1,4-diyl dianion. Tetrahedron Lett. 1988, 29, 6939–6942. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS; Empirical Absorption Correction Program; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXL-2014/7; Program for the Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Cpd | d/Å | Angle Sum at Si/° | β/° | τ/° | δ(29Si) 1 | λmax/nm (ε) 2 |
|---|---|---|---|---|---|---|
| 1 | 2.1762(5) | 358.52(3) (Si1–SiMe3), 359.61(3) (Si2–SiMe3) | 12.9 (Si1–SiMe3), 6.5 (Si2–SiMe3) | 17.7 | 131.4 (=Si–SiMe3) | 420 (1.57 × 104), 343 (4.63 × 103) |
| 4 | 2.2035(5) | 358.04(3) (=Si···K), 359.94(3) (=Si–SiMe3) | 3.7 (=Si···K) 3, 1.5 (=Si–SiMe3) 3 | 2.5 | 146.9 (=Si–SiMe3) 219.3 (=Si−) | – |
| 5 | 2.1860(19) | 358.8(1) (=Si–SiEt3), 358.6(1) (=Si–SiMe3) | 11.2 (=Si–SiEt3), 12.1 (=Si–SiMe3) | 17.6 | 123.5 (=Si–SiEt3), 134.2 (=Si–SiMe3) | – |
| 6 | 2.1598(6) | 360.00(5) (=Si–Ant), 355.98(3) (=Si–SiMe3) | 0.1 (=Si–Ant), 22.5 (=Si–SiMe3) | 0.1 | 130.3 (=Si–Ant), 74.9 (=Si–SiMe3) | 535 (9.25 × 102), 399 (2.64 × 104) |
| 6Br | 2.1711(7) | 359.76(6) (=Si–AntBr), 356.04(5) (=Si–SiMe3) | 4.7 (=Si–AntBr), 22.4 (=Si–SiMe3) | 8.5 | 75.1 (=Si–SiMe3), 129.1 (=Si–AntBr) | [578 (9.67 × 102), 410 (3.08 × 104)] 5 |
| 7 | 2.1525(7) | 357.10(6) (Si1–Ant), 355.71(6) (Si2–Ant) | 17.4 (=Si1–Ant), 21.0 (=Si2–Ant) | 3.3 | 88.2 (=Si–Ant) | 543 (1.68 × 103) 399 (1.85 × 104) |
| 8 4 | 2.1850(4) (Si1=Si2) 2.1915(5) (Si3=Si4) | 359.99(4) (Si1), 359.99(3) (Si2) 359.98(3) (Si3) 359.94(4) (Si4) | 0.3 (Si1) 1.2 (Si2) 1.0 (Si3) 2.4 (Si4) | 0.7 (Si1=Si2) 1.8 (Si3=Si4) | 106.4 (Si=SiSiMe3), 143.0 (Si=SiSiMe3) | ~500 (sh, 3.57 × 103), 437 (3.42 × 104) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akasaka, N.; Tanaka, K.; Ishida, S.; Iwamoto, T. Synthesis and Functionalization of a 1,2-Bis(trimethylsilyl)-1,2-disilacyclohexene That Can Serve as a Unit of cis-1,2-Dialkyldisilene. Inorganics 2018, 6, 21. https://doi.org/10.3390/inorganics6010021
Akasaka N, Tanaka K, Ishida S, Iwamoto T. Synthesis and Functionalization of a 1,2-Bis(trimethylsilyl)-1,2-disilacyclohexene That Can Serve as a Unit of cis-1,2-Dialkyldisilene. Inorganics. 2018; 6(1):21. https://doi.org/10.3390/inorganics6010021
Chicago/Turabian StyleAkasaka, Naohiko, Kaho Tanaka, Shintaro Ishida, and Takeaki Iwamoto. 2018. "Synthesis and Functionalization of a 1,2-Bis(trimethylsilyl)-1,2-disilacyclohexene That Can Serve as a Unit of cis-1,2-Dialkyldisilene" Inorganics 6, no. 1: 21. https://doi.org/10.3390/inorganics6010021
APA StyleAkasaka, N., Tanaka, K., Ishida, S., & Iwamoto, T. (2018). Synthesis and Functionalization of a 1,2-Bis(trimethylsilyl)-1,2-disilacyclohexene That Can Serve as a Unit of cis-1,2-Dialkyldisilene. Inorganics, 6(1), 21. https://doi.org/10.3390/inorganics6010021