2.1. Crystal Structure and Bonding in Ba4(Li1−xGax)7Sb6, Ba4(Li1−xInx)7Sb6, Ba4(Li1−xInx)7Bi6, and Eu4(Li1−xInx)7Bi6
All the compounds of the “4-7-6” variety crystallize in the monoclinic space group
C2/
m with their own structure type [
13]. There are a total of 34 atoms in the unit cell and two formula units, thus, the Pearson symbol of this structure is
mS34.
A ternary compound with this structure was reported by us in 2014, namely Eu
4Li
7Bi
6 [
13]. At that time, the structure was considered unique, although we noted the existence of the phase known as Ba
8Li
13GaSb
12, reported eight years earlier [
17]. Because the latter was worked out in non-standard coordinate settings (the published Wyckoff sequence of the structure is
i8
d) [
17], ICSD references it as isotypic to the Ce
4Mg
7Ge
6-type structure [
18]. Our analysis from 2014 of the bonding in Eu
4Li
7Bi
6 [
13] and Ce
4Mg
7Ge
6 [
18] indicated that although both have the same
C2/
m space group and same number of atoms in the unit cell, the structures are sufficiently different and should not be considered isotypic. This conjecture is also supported by the standardized version of the coordinates, which shows that Eu
4Li
7Bi
6 and its herein presented analogs have an
i8
c Wyckoff sequence. Furthermore, the refined structure of Ba
4(Li
1−xGa
x)
7Sb
6 (
x ≈ 0.08, i.e., Ba
4(Li
0.92Ga
0.08)
7Sb
6 = Ba
4Li
~6.4Ga
~0.6Sb
6) shows that the previously reported Ba
8Li
13GaSb
12 (=Ba
4Li
6.5Ga
0.5Sb
6) has the same structure as all other members of the “4-7-6” family of pnictides (
Table 1,
Table 2 and
Table 3), and that the structures of the ternary rare-earth metal–magnesium germanides belonging to the Ce
4Mg
7Ge
6 structure type are related, but not identical.
Crystallographic data for several crystals from each of the Ba
4(Li
1−xIn
x)
7Sb
6 and Ba
4(Li
1−xIn
x)
7Bi
6 structures are listed in
Table 2. The analogous information from two Eu
4(Li
1−xIn
x)
7Bi
6 crystals is given in
Table 3. In all cases, the refined parameters included the scaling factors, atomic coordinates, displacement parameters, and where applicable—the extinction coefficient and occupancy factors. Relevant interatomic distances in the Ba
4(Li
1−xGa
x)
7Sb
6, Ba
4(Li
1−xIn
x)
7Sb
6, Ba
4(Li
1−xIn
x)
7Bi
6, and Eu
4(Li
1−xIn
x)
7Bi
6 structures are displayed in
Table 4. Points of specific interest will be discussed in detail later, here, but we just mention that all of the
AE–
Pn,
Pn–
Pn, and Li–
Pn distances are distributed between 3.6487(6)–3.3394(6) Å, 3.0589(9)–2.8472(10) Å, and 3.4407(4)–2.799(9) Å, respectively. These metrics are consistent with those reported for CaLiSb, BaLiSb, Ba
3Li
4Sb
4, SrLiBi, Ba
3Li
4Bi
4, and Eu
4Li
7Bi
6 [
13,
19,
20,
21,
22,
23].
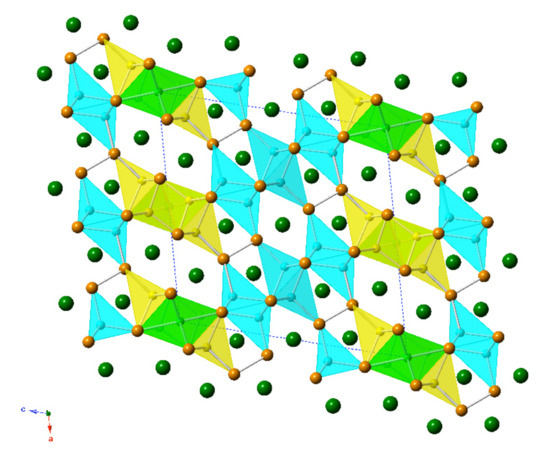
The overall structure is schematically shown in
Figure 1. As seen in the structural representation, there are different coordination polyhedra. Li1, Li2, and Li3 atoms are all tetrahedrally coordinated by Sb, while Li4 is octahedrally coordinated. The polyhedra interact through edge and corner sharing. Naturally, because of the arrangement of these fused polyhedra, some relatively short Li–Li contacts arise (Li1–Li3 3.05(1) Å, Li1–Li4 2.98(3) Å). However, considering the Pauling single-bonded radius,
rLi = 1.225 Å [
24], such distances are not indicative of strong bonding interactions.
Pn1 atoms form [
Pn2]
4− dimers (isoelectronic with the I
2 molecule), while
Pn2 and
Pn3 atoms exist as isolated
Pn3− ions. In the tetrahedra where Li is mixed and occupied with Ga or In, the distances to the closest neighbors vary from 2.867(5) to 3.071(2) Å. Note also that the Sb–Sb bond lengths are almost invariant of other structural parameters, which is indicative of strong covalent bonds; they vary in magnitude from 2.8427(13) to 2.8472(10) Å, and they are shorter than the 2.908 Å Sb–Sb distance found in elemental Sb [
25]. The same can be said for the Bi–Bi bonding too, where the Bi–Bi distances are in the range 3.0443(8) to 3.0522(10) Å, which is shorter than the distances in elemental Bi [
26]. However, this finding is not surprising, as similar values have been reported for KSb, Ba
3Li
4Bi
4, Sr
3Li
4Sb
4, Eu
3Li
4Sb
4, KBi, SrLiBi, Ba
21Cd
4Bi
18, and Eu
4Li
7Bi
6 [
13,
20,
27,
28,
29]. The Ba atoms prefer sites with high coordination numbers, and they can be viewed as Ba
2+ cations residing within channels of the [Li
7Sb
6] and [Li
7Bi
6] polyanionic sub-structure (exaggerating of course the covalency of the Li–Sb and Li–Bi bonds).
We also draw attention to the Eu
4Li
6.60In
0.40(1)Bi
6 structure and that of Eu
4Li
7Bi
6 as reported by our group in a previous study [
13]. Rigorous assessment of the unit cell parameter and the interatomic distances show very clear differences, which attest to the structural response to the variation of the electron count (notice that the ionic radii of Li
+ and In
3+ for the same coordination number 4 are very close, 0.59 and 0.62 Å, respectively [
30], and geometric factors will not be expected to play a big role, especially at this level or with Li–In substitution). First, let us compare the unit cell parameters and volumes. They are the following:
a = 17.558(3) Å,
b = 4.8114(8) Å,
c = 12.812(2) Å, β =126.035(2)°,
V = 875.2(3) Å
3 for Eu
4Li
7Bi
6, and
a = 17.607(3) Å,
b = 4.8222(8) Å,
c = 12.826(2) Å, β =125.929(2)°,
V = 881.8(2) Å
3) for Eu
4Li
6.60In
0.40(1)Bi
6. The ca. 1% increase in the unit cell volume correlates well with some subtle changes in the bond distances and bond angles.
Overall, all bonds are slightly lengthened in the structure with the larger cell volume, Eu4Li6.60In0.40(1)Bi6, but identifying the most affected bonds is challenging, as many of the changes are within 3–5 e.s.d.s. For example, the Bi2 dumbbells elongate by 0.006(1) Å. Li–Bi interactions in the tetrahedral coordination are the ones that appear to show some statistically significant changes, which are mostly evident from the changes in bond angles. Focusing on the Li3 site, where the In substitution occurs, one can notice that while dLi3−Bi3 = 2.866(4) Å/dLi3−Bi2 = 2.990(3) Å in Eu4Li6.60In0.40(1)Bi6 are similar to dLi3−Bi3 = 2.86(3) Å/dLi3−Bi2 = 2.92(1) Å in Eu4Li7Bi6, the Bi2–Li3–Bi2 bond angles are slightly different, 107.5(1)° for the former structure and 111.0(8)° for the latter, meaning a slightly different type of tetrahedral distortion in either case.
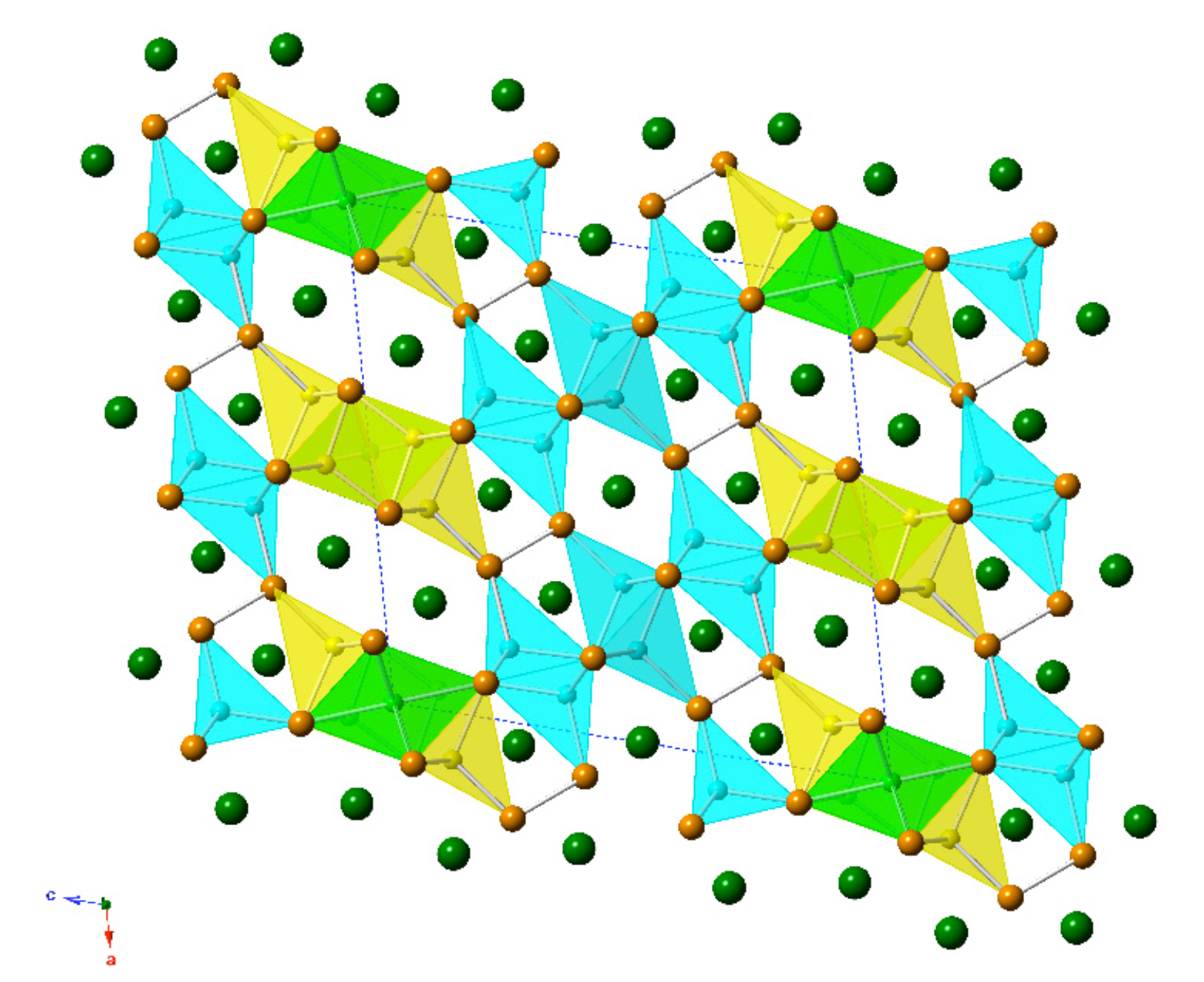
2.2. Crystal Structure and Bonding in Ba7Li11Bi10
The new compound, Ba
7Li
11Bi
10, was discovered and synthesized for the first time from stoichiometric reactions of the elements as part of this study. Ba
7Li
11Bi
10 crystallizes with its own structure type, the Pearson index
mS56 (
Table 5). It is structurally related to the aforementioned “4-7-6” structures, which have 9 independent atomic positions (four for alkaline/rare-earth metals, three for the pnictogen atoms, and four for lithium atoms) in the asymmetric unit, while the Ba
7Li
11Bi
10 structure has 15 symmetry-unique positions (four for barium atoms, five for the bismuth atoms, and six for lithium atoms) in the asymmetric unit. The Wyckoff sequence for this new crystallographic arrangement is
i13
d a. Consequently, the structure is rather complex (
Figure 2). A common trait is shared between the Ba
4(Li
1−xIn
x)
7Bi
6 and Ba
7Li
11Bi
10 structures. They both have anionic substructures based on isolated Bi
3− ions and [Bi
2]
4− dimers. The positions of the Li atoms in the two structures are such that the LiBi
4 tetrahedra have similar connectivity, and the interatomic distances are closely matching (
Table 4 and
Table 6).
The observed average Li–Bi distances range between 2.799(9) Å to 3.071(2) Å, and 2.96(3) Å to 3.10(2) Å, for the Ba
4(Li
1−xIn
x)
7Bi
6 and Ba
7Li
11Bi
10 structures, respectively. These values are in close range with the sum of the Pauling covalent radii;
rLi +
rBi = 1.225 Å + 1.510 Å [
24], as well as with those reported for other compounds [
13,
20,
27,
28,
29]. The tetrahedra are slightly distorted in both structures with the values for the Bi1–Li1–Bi2/Bi1–Li2–Bi3 bond angles ranging between 104.7(4)° and 111.8(3)°.
Similar arguments used above can be applied to the LiBi6 octahedra, where the average Li–Bi bond distances in the Ba4(Li1−xInx)7Bi6 and Ba7Li11Bi10 structures range between 3.4419(5) Å and 3.4407(4) Å for the former, and between 3.4274(6) Å and 3.4398(8) Å for the latter. In general, the bond lengths in the octahedra are longer than those in tetrahedral, as expected. The lengths of the Bi–Bi bonds in the Ba4(Li1−xInx)7Bi6 and Ba7Li11Bi10 structures match within two e.s.d.s.
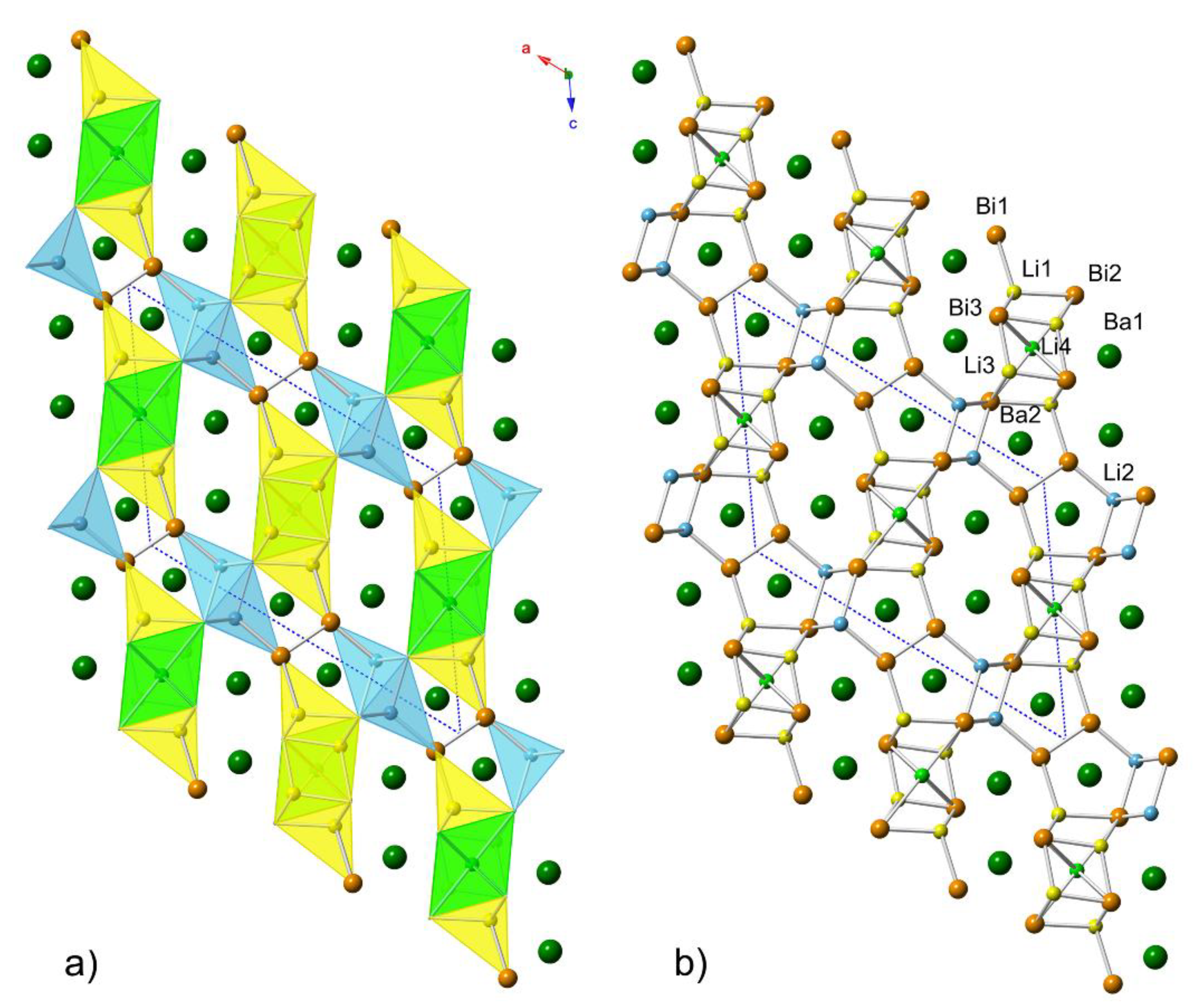
2.3. Structural Relationships
Considering the similarities between the “4-7-6” and “7-11-10” structures, it can be proposed that the two are members of a homologous series. Let us illustrate this idea by taking two compounds with known structures—Ba
3Li
4Bi
4 (Zr
3Cu
4Si
4 structure type, Pearson symbol
oI44) and BaLi
3Bi
2 (LaLi
3Sb
2 structure type, filled version of the CaAl
2Si
2 structure type, Pearson symbol
hP6). We draw attention to the fact that the existence of the former compound has been experimentally confirmed, but the latter is not known. For electronic stability reasons, as discussed in the next section, BaLi
3Bi
2 is not likely to form, and its closest analog would be EuLi
2.58In
0.42(2)Bi
2, which was obtained as a part of this study (see the
Supplementary Materials). For comparison, the isotypic lanthanide-based
RELi
3Bi
2 are valance-precise semiconductors, whose formulae can be represented as (
RE3+)(Li
1+)
3(Bi
3−)
2, and they can be readily made [
31]). Nonetheless, for the purposes of this discussion, BaLi
3Bi
2, even as a hypothetical case, will be considered, and slabs cut out from Ba
3Li
4Bi
4 and BaLi
3Bi
2 can be integrated to form Ba
4Li
7Bi
6 following Equation (1):
A detailed description of the structural relationships that equation 1 describes is already discussed in the literature [
13]. Now, using the same two fragments, the structure of Ba
7Li
11Bi
10 can be seen as realized through Equation (2):
In other words, Ba
4Li
7Bi
6 and Ba
7Li
11Bi
10 are the simplest members of the family described with the general formula (BaLi
3Bi
2)
n(Ba
3Li
4Bi
4)
m, where the Ba
4Li
7Bi
6 is the member with
n = 1 and
m = 1, and Ba
7Li
11Bi
10 is the intergrowth of the two components with
n = 1 and
m = 2, respectively. A pictorial representation of this analogy is shown in
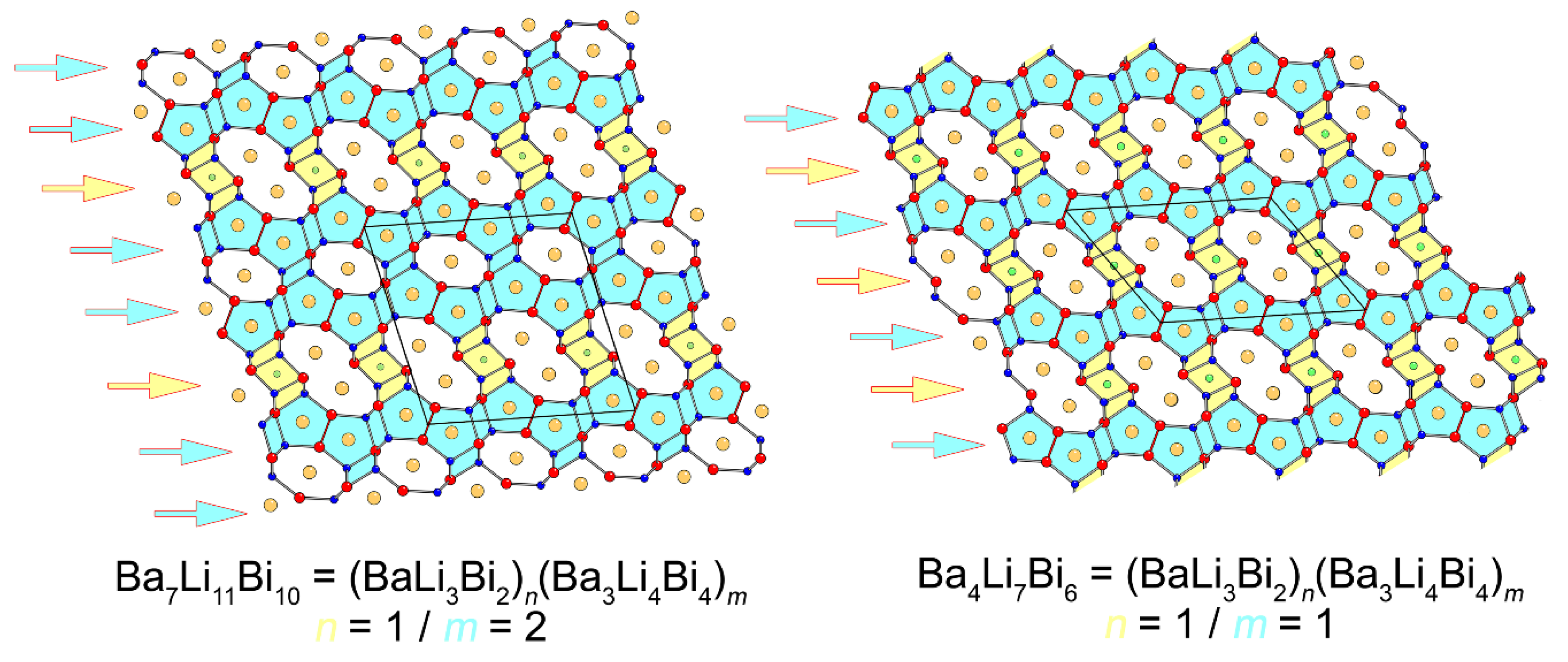
Figure 3.
The illustrated idea allows for the prediction of other possible members. For example, switching the ratio, i.e., taking the two components with n = 2 and m = 1, leads to Ba5Li10Bi8 = (BaLi3Bi2)2(Ba3Li4Bi4)1. Going above and beyond, based on this, one can envision the existence of other higher-order analogs such as Ba6Li13Bi10 (n = 3 and m = 1) or Ba10Li15Bi14 (n = 1 and m = 3). Experiments aimed at making these compounds were attempted, but so far they have been unsuccessful.
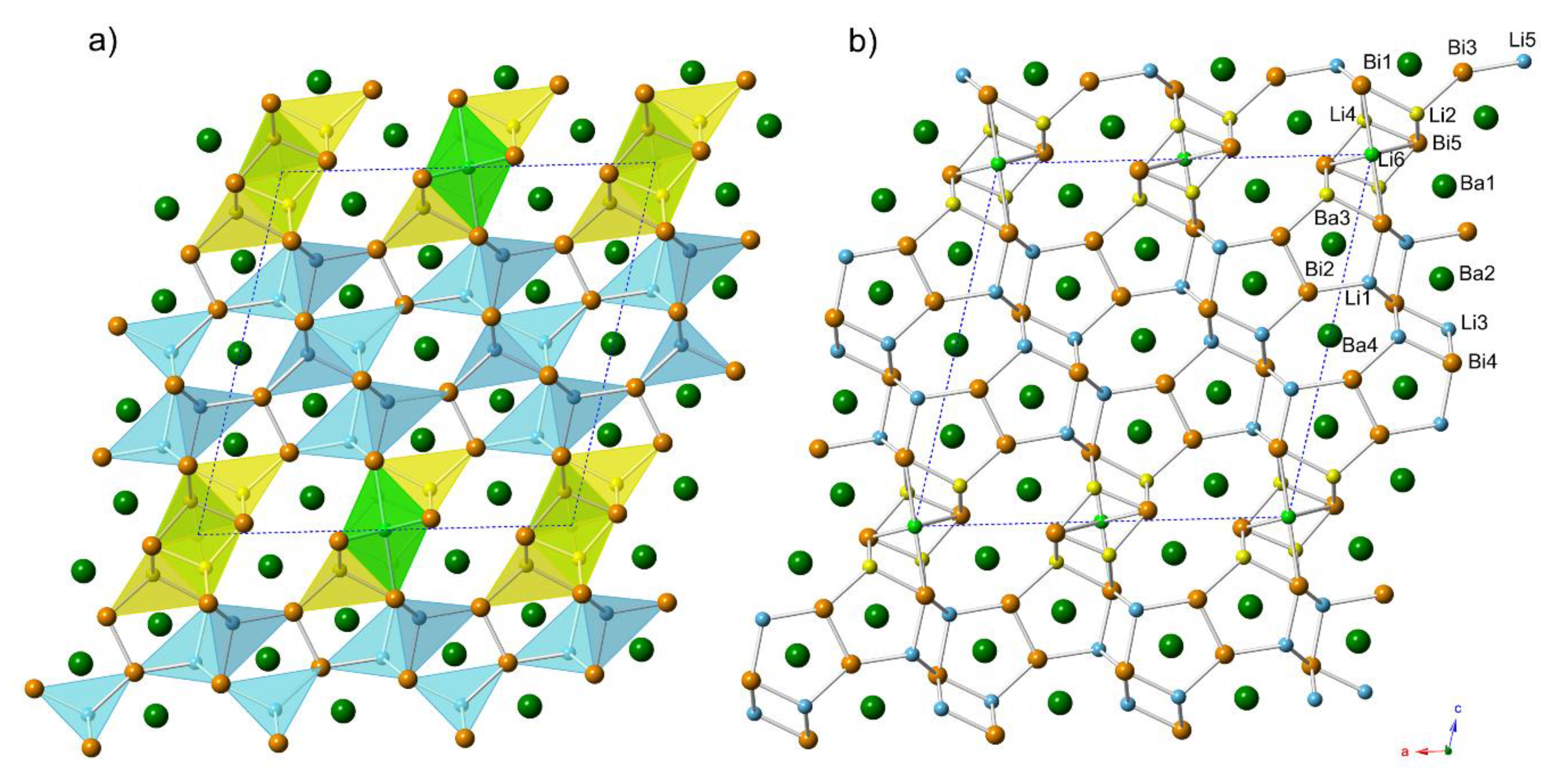
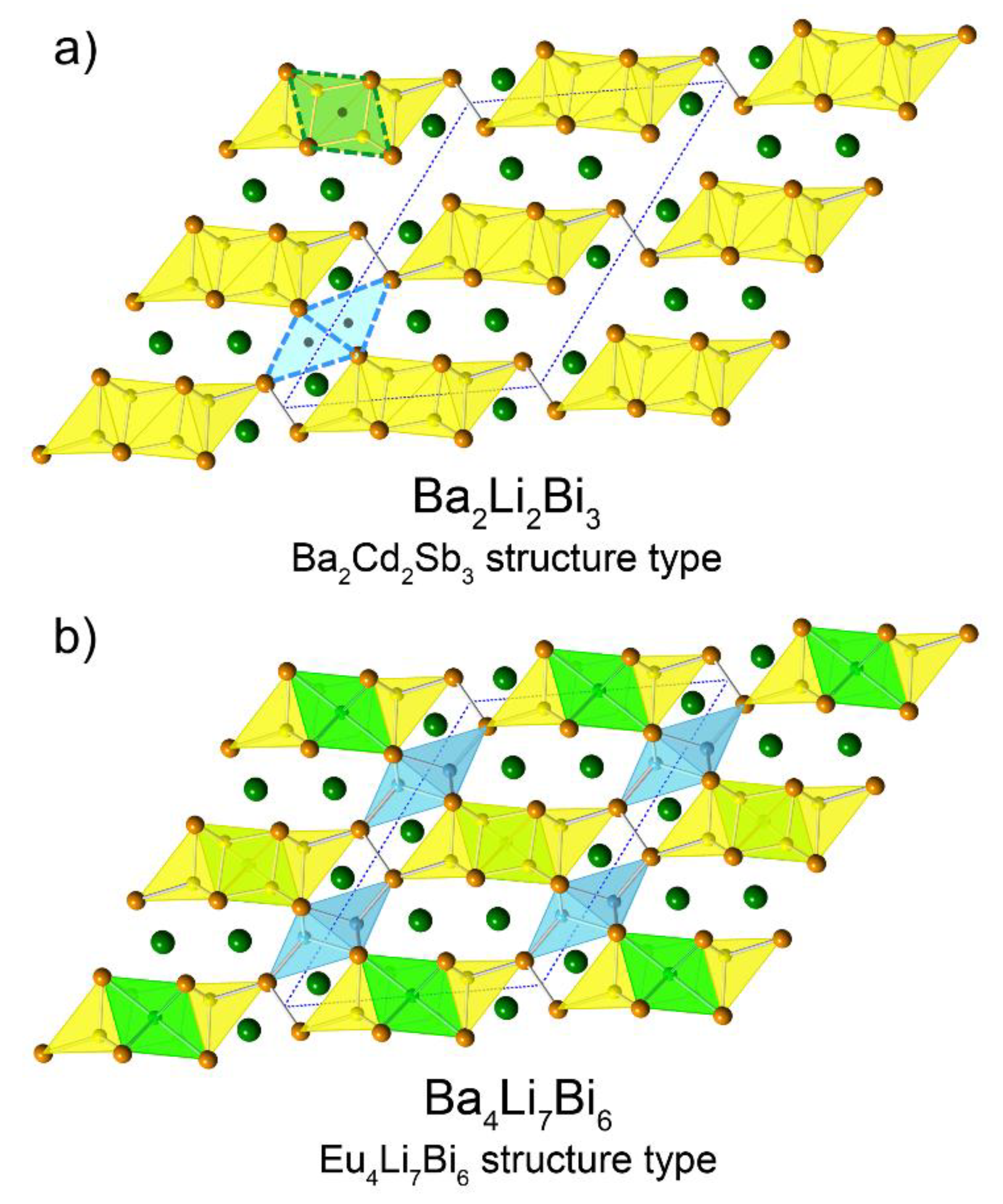
Another interesting structural relationship that can be mentioned here concerns the similarities between the structure of Ba
4Li
7Bi
6 and that of Ba
4Cd
4Bi
6 (=Ba
2Cd
2Bi
3), which is actually not reported yet, but presumed to be isotypic with Ba
2Cd
2Sb
3 (own type with the monoclinic space group
C2/
m and Pearson symbol
mS28) [
32]. The latter structure is described as a derivative of BaCd
2Sb
2 with the CaAl
2Si
2 structure type. Recall that the BaLi
3Bi
2 compound that we considered in our homologous series is the same basic structure, where an extra Li atom is inserted within. Given that Cd is nominally divalent, one can reason that the very same structure that is adopted by the Cd-bearing compound cannot be realized if Li (nominally monovalent) is used. Instead, the Ba
2Li
2Bi
3 sub-structure can be seen as being distorted a little around the pivotal Bi
2-dimers, which creates two more tetrahedral holes that are suitable for Li (described by one crystallographic position, Li2 in the notation used for the “4-7-6” structures—
Figure 1). Still two electrons short of the ideal number of valence electrons, the imaginary Ba
4Li
6Bi
6 accepts one more Li atom, in an octahedral hole (described by position Li4 in the notation used for the “4-7-6” structures). A pictorial representation of this analogy is shown in
Figure 4.
2.4. Valence Electron Count and Electronic Band Structure
As mentioned earlier in this paper, Eu
4Li
7Bi
6 does not appear to satisfy the Zintl–Klemm electron counting scheme [
33], as attempts to rationalize the structure ((Eu
2+)
4(Li
+)
7([Bi
2]
4−)(Bi
3−)
4(
h+), where
h+ denotes an electron hole) fail to produce a charge-balanced formula [
13]. In other words, the compound Eu
4Li
7Bi
6, joins the ranks of electron-deficient “near” Zintl phases, such as the families of Ca
14MnBi
11 [
34] and Ca
9Zn
4Sb
9 [
35]. The structure of the former has been experimentally confirmed to contain an electron hole, and it can be judiciously tuned to a Zintl phase by the aliovalent substitution of one Ca
2+ cation by one
RE3+ cation (
RE = rare-earth metal), as demonstrated with the examples of Ca
13REMnBi
11 [
36] and Ca
13REMnSb
11 [
37]. On the other hand, the originally published Ca
9Zn
4Sb
9 structure has been revisited, and an interstitial position within it has been identified, i.e., the formula is Ca
9Zn
4+xSb
9 [
38], which also brings the valence electron count very close to the expected from the Zintl concept. This structure has also been shown to be amenable to tuning via substitution, as demonstrated with the example of Ca
8REMn
4Sb
9 [
39], where the interstitial site is empty, but the electron count is modulated by the extra electron contributed by the rare-earth metal substituting for Ca.
On the opposite side of the spectrum, here are some nominally Zintl phases with the “wrong” electron count. Good examples are the structures of Ca
4Bi
2O (previously thought to be Ca
2Bi) [
40], and the unusual Ba
5Cd
2Sb
5O
0.5 [
41]. For both of them, the perceived electron surplus has helped to identify the unrecognized oxygen impurity. Considering the ambivalent role that Li can play in intermetallics, we might bring up the case studies carried out on germanides/stannide with complex structures, such as
AELi
1−xIn
xGe
2,
RE4Li
4−xSn
4+x and Li
9−xEuSn
6+x [
14,
15,
16], which show the tendency of such structures to attain more optimal valence electron counts by Li-group 13 or Li-group 14 element substitutions.
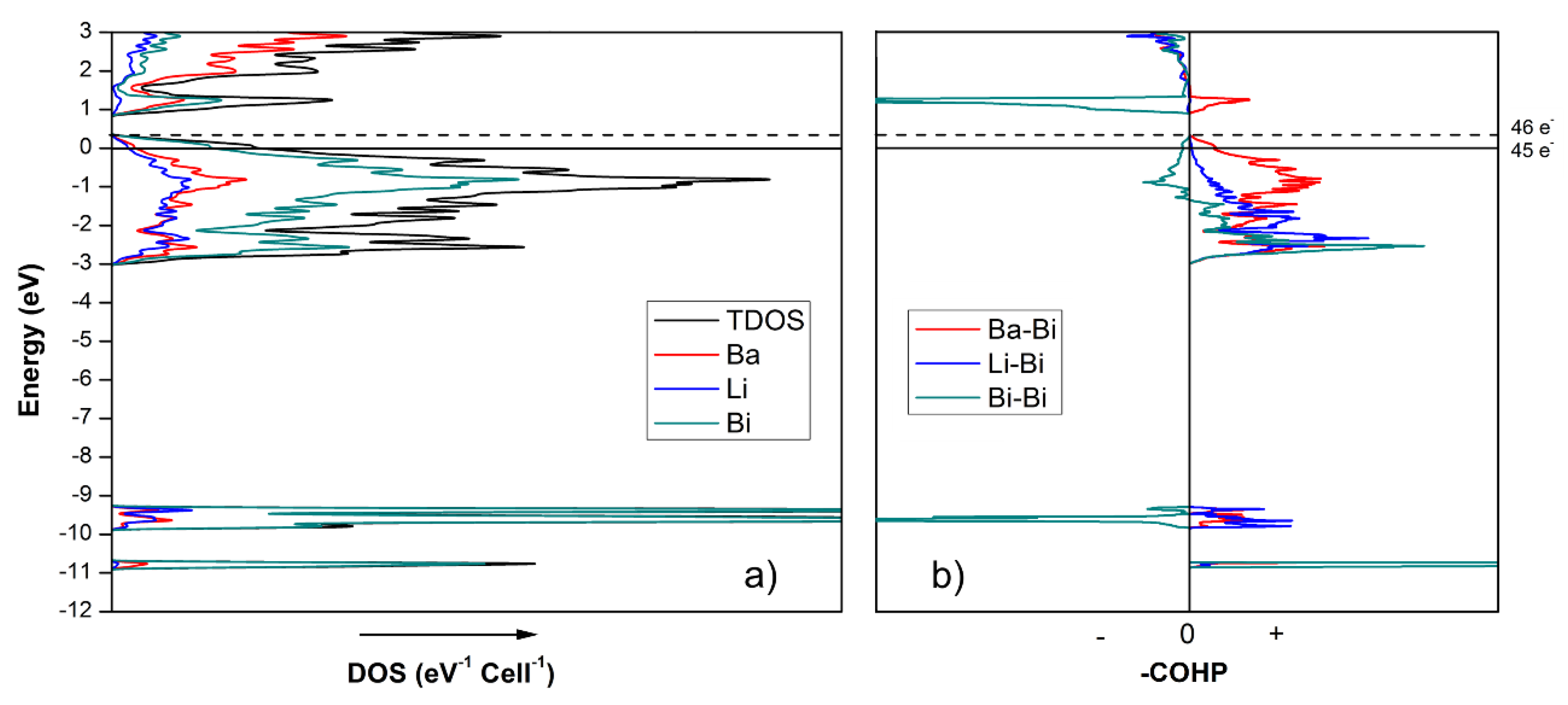
Armed with the knowledge from the above-mentioned studies, we considered many different hypotheses, to ultimately settle on the notion that the electron count for the “4-7-6” compounds needs to be augmented, which is achieved by virtue of substituting a fraction of the Li atoms with either Ga or In atoms. As seen from the total and partial density of states (DOS) curves of Ba
4Li
7Bi
6 (
Figure 5), the integrated DOS at the Fermi level (
EF = 0 eV) corresponds to 45 valence electrons per unit cell. Note that the calculation is done on Ba
4Li
7Bi
6, which is an ordered model of the actual Ba
4(Li
1−xIn
x)
7Bi
6 structure, where the mixed occupied Li/In site, according to the single-crystal structure refinement data, were treated as only occupied by Li atoms. A closer inspection shows that an additional electron is required to reach the top of the valence band, in analogy to the findings previously discussed for Eu
4Li
7Bi
6 [
13]. Thus, the electronic calculations are in excellent agreement with the formulation based on the Zintl–Klemm concept. Given the inability to prepare other pure ternary compounds with this structure with Ba or Sr, both of which can contribute only two electrons to the bonding, it can be surmised that the hypothetical Ba
4Li
7Bi
6 cannot be realized, and that Ga or In doping on the Li site is critical to obtain more electronically stable, charge-balanced compounds.
Ba
7Li
11Bi
10 is not an electron-precise compound either. According to Zintl concept, the empirical formula of Ba
7Li
11Bi
10 can be broken down to (Ba
2+)
7(Li
+)
11([Bi
2]
4−)
2(Bi
3−)
6(
h+), which indicates a shortage of one electron per formula unit. This is not surprising, given that the building blocks of Ba
4Li
7Bi
6 and Ba
7Li
11Bi
10 are the same (
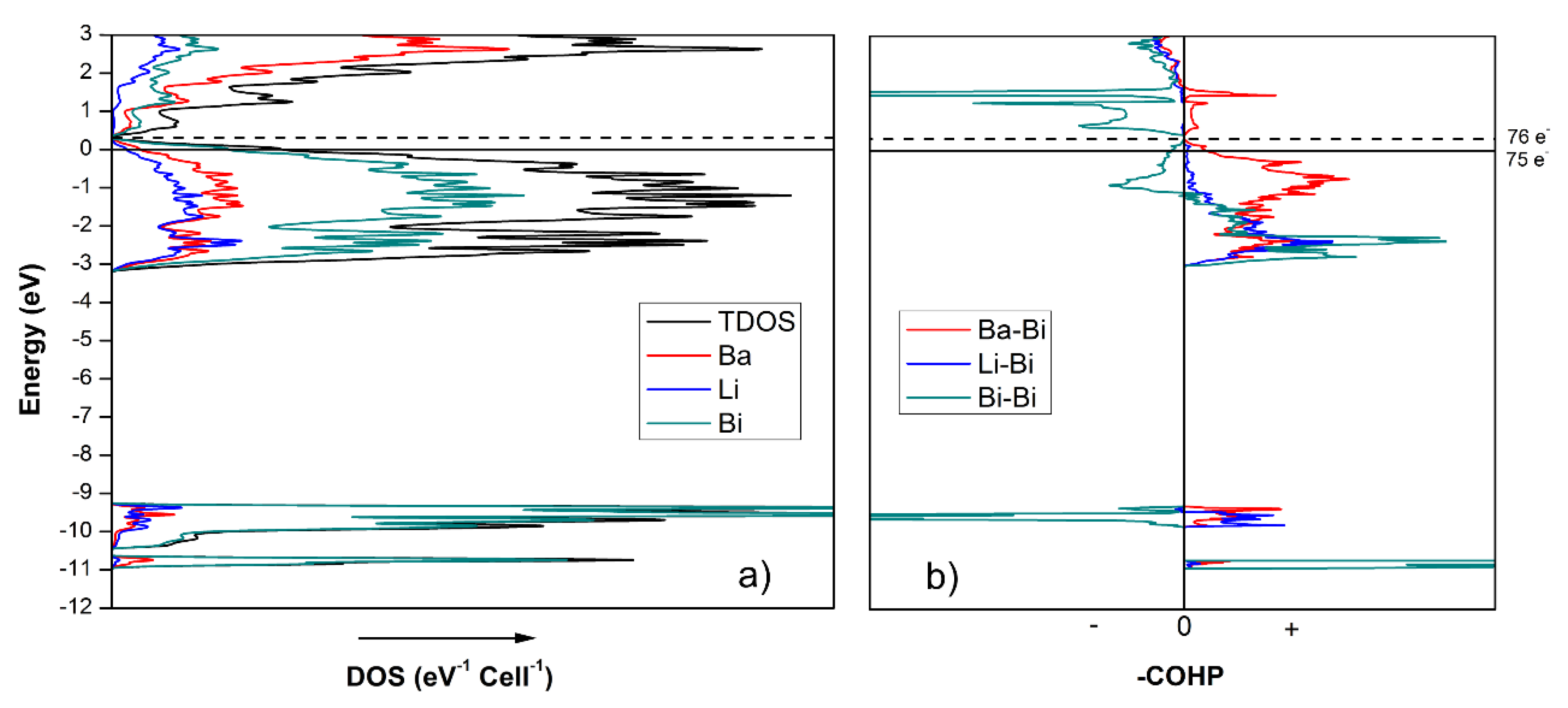
vide supra). The total and partial density of states (DOS) curves of Ba
7Li
11Bi
10 are shown in
Figure 6, and agree with this conjecture—indeed, similar electronic characteristics can be noticed. For instance, both structures have non-zero DOS at the Fermi level, indicating metallic behavior. The
s orbitals of Bi contribute almost exclusively to the energy DOS peaks in the −11 to −9.5 eV range, with the majority of them being lone pairs. Considering the very sharp bonding and antibonding peaks in the same energy range, as shown in the crystal orbital Hamilton population (COHP) in
Figure 6b, the
s orbitals of Bi are also responsible for the σ-bonding (and σ*-antibonding) states of the Bi
2-dimer. The
p orbitals of Bi are mixed with Ba and Li orbitals in the range between −3 eV to the Fermi level, suggesting the emergence of multiple covalent type interactions between these atoms.
The integrated DOS for Ba
7Li
11Bi
10 (
Figure 6) at
EF = 0 eV corresponds to 75 valence electrons per unit cell. If the rigid band model is employed, integrating the DOS to 76 valence electrons, which is what the Zintl concept predicts, moves the Fermi level up in energy to a local DOS minimum at ca. 0.3 eV. This is perhaps the most notable difference between the electronic structures of Ba
4Li
7Bi
6 and Ba
7Li
11Bi
10, where an augmented electron count for the former leads to an electronic structure that is fully optimized, and a sizeable band-gap is opened, whereas for the latter, adding one more electron only helps to reach a nearly vanishing energy gap. This small difference might explain why doping in the Ba
7Li
11Bi
10 structure is not as critical as it is for the Ba
4Li
7Bi
6 counterpart, in line with the realization that, as discussed earlier, the Ba
4Li
7Bi
6 structure might be seen as being made up of 50% BaLi
3Bi
2-fragments, which are electron-deficient by nature. By comparison, in Ba
7Li
11Bi
10, the contribution of the electron-deficient BaLi
3Bi
2-component is reduced to one-third.
For a better understanding of the atomic interactions, the crystal orbital Hamilton population (COHP) curves were constructed and closely examined. In general, the bonding picture in herein is similar to what was previously described for the Eu
4Li
7Bi
6 compound [
13]. The bonding description here is valid for both Ba
4Li
7Bi
6 and Ba
7Li
11Bi
10 compounds. The COHP curve of the Bi–Bi dimers show antibonding character close to the Fermi level in addition to some deeper bonding states. For both structures, the values for the integrated –COHP for Bi–Bi interactions are essentially the same at the Fermi level (~1.6 eV per bond). Although a direct comparison between the bonding strengths of different linear muffin-tin orbital (LMTO) calculations is not valid, we can see that the magnitude of the Bi–Bi bonding interactions is approximately the same. Judging from the –COHPs, the antibonding π* and σ* states of the [Bi
2] dimers derived from p-orbitals are not completely occupied, which may indicate that the bond order within the dimers is not 1, like in the I
2 molecule to which an analogy was drawn earlier, but higher. The notion of a “double bond” is inconsistent with the lengths of the Bi–Bi bonds in both Ba
4(Li
1−xIn
x)
7Bi
6 and Ba
7Li
11Bi
10 structures (
Table 4 and
Table 6, respectively), but partial/non-integer bond order is conceivable. Thus, the formulae Ba
4Li
7Bi
6 and Ba
7Li
11Bi
10 can be formally represented as (Ba
2+)
4(Li
+)
7([Bi
2]
3−(Bi
3−)
4 and (Ba
2+)
7(Li
+)
11([Bi
2]
3.5−)
2(Bi
3−)
6, respectively. In this view, the idealized “4-7-6” compounds will have a higher bond order (i.e., 1.5) tan the “7-11-10” ones, but this difference is offset in the actual “4-7-6” compounds by the partial substitution of In or Ga for Li; hence, the formal charge of the [Bi
2] moieties in both is about the same.
The Li–Bi bonds are optimized, reflecting the strong covalent nature, while the Ba–Bi interactions are slightly underoptimized (electron-deficient) because there are still bonding states available above the Fermi level. This situation will not change even by adding more electrons, i.e., moving the Fermi level up to the gap. This scenario is not unusual for highly polar interactions with strong ionic contribution [
42]. Similarly, the Li–Bi will not change much if the Fermi level were shifted up because only a few states are available above the Fermi level and are of anti-bonding character for the Bi–Bi interactions and bonding for Li–Bi ones. We can estimate that for both structures, the integrated –COHP values for the Li–Bi interactions lie between 0.1 and 0.4 eV per bond, while for the Ba–Bi interactions, they range between 0.4 and 0.7 eV per bond.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}