Solvent and Substituent Effects on the Phosphine + CO2 Reaction





Abstract
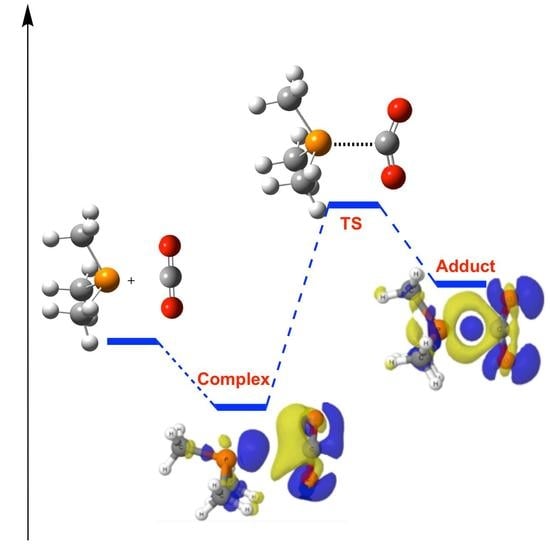
:
1. Introduction
2. Results
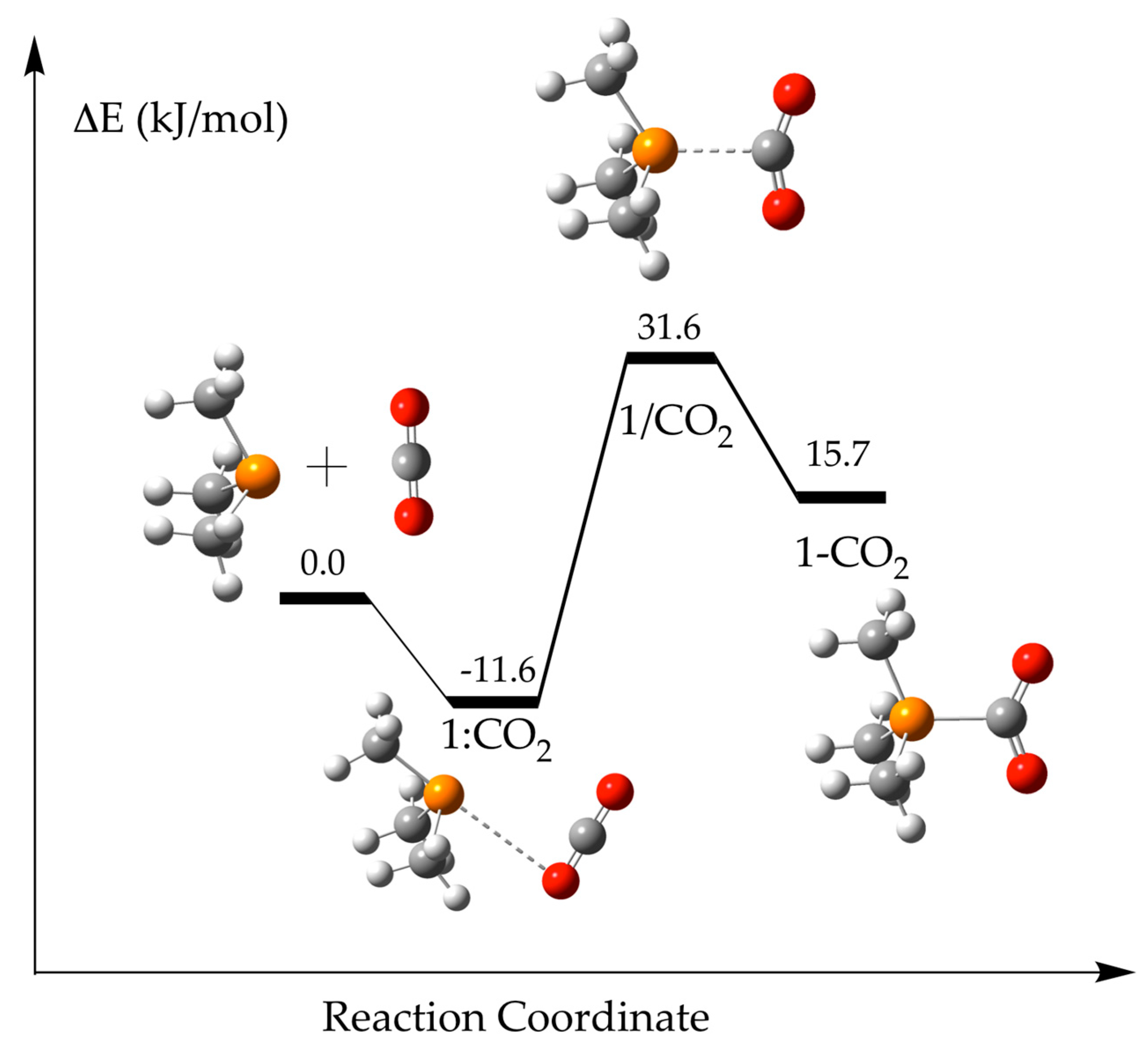
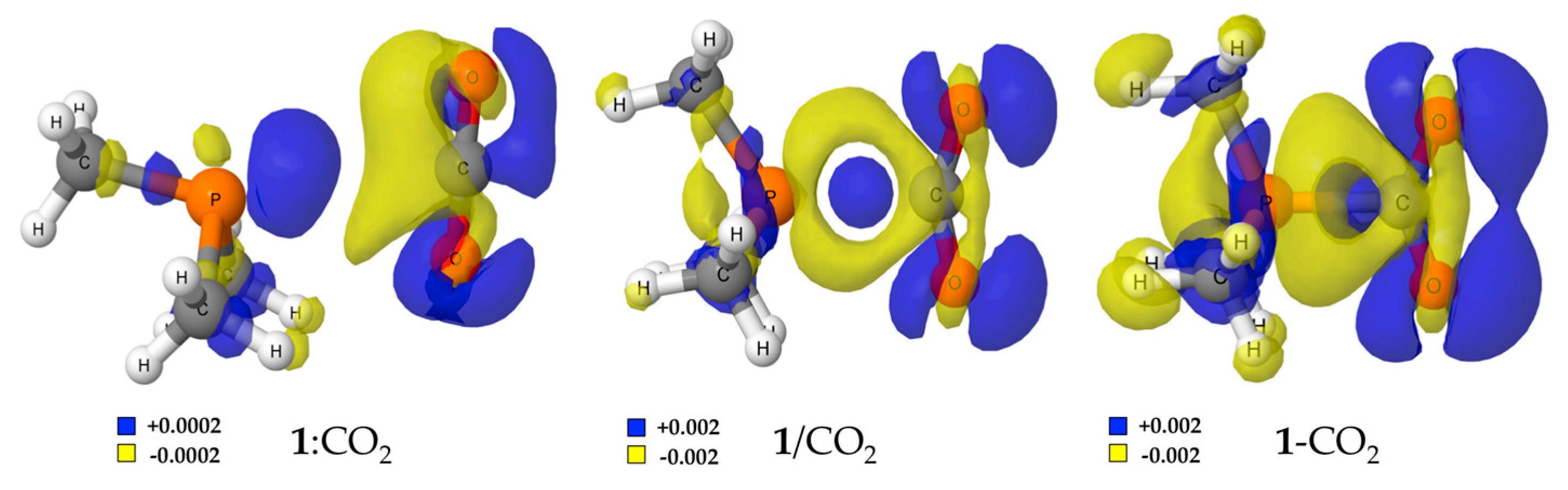
2.1. (CH3)3P + CO2 (1)
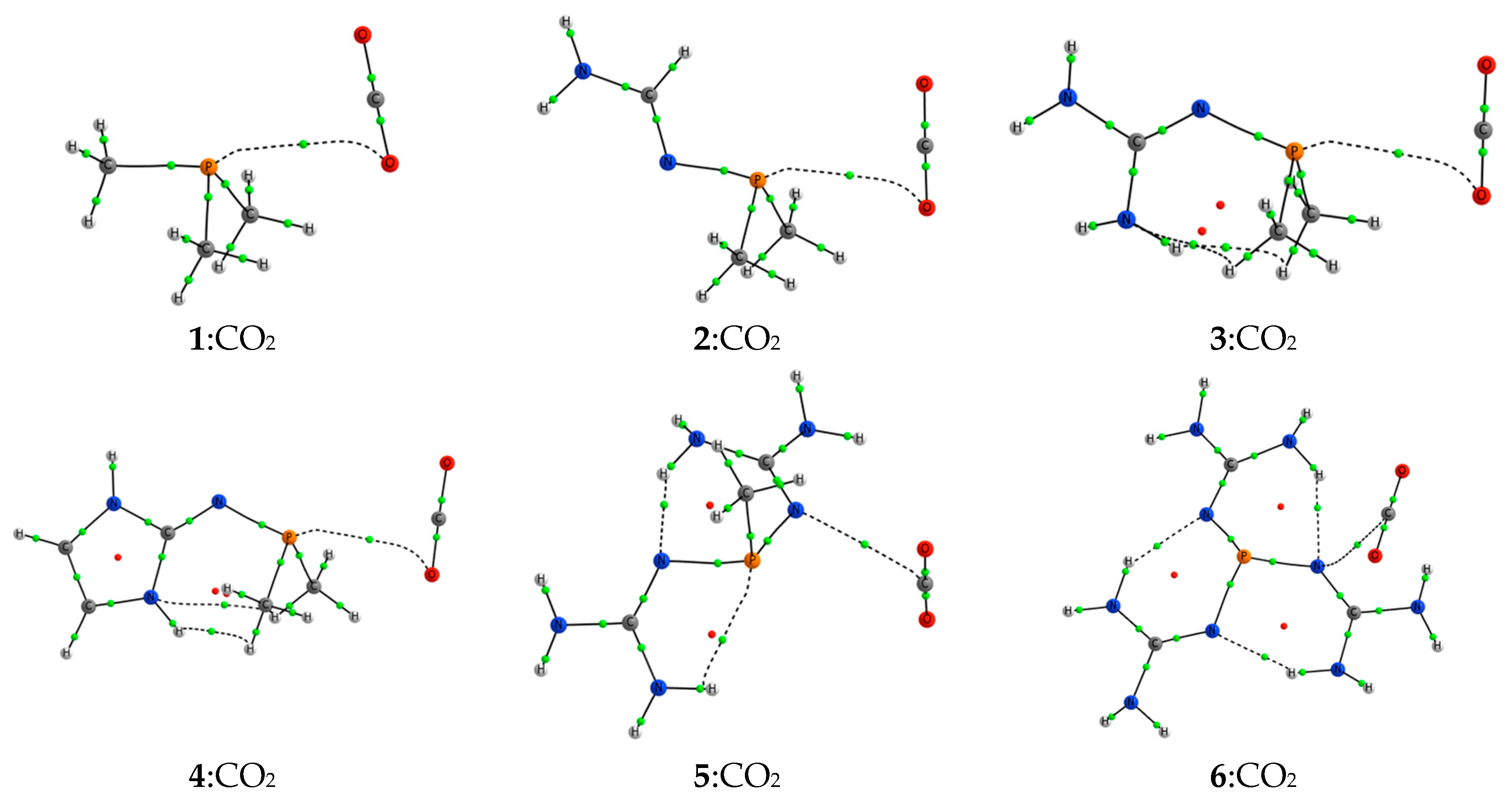
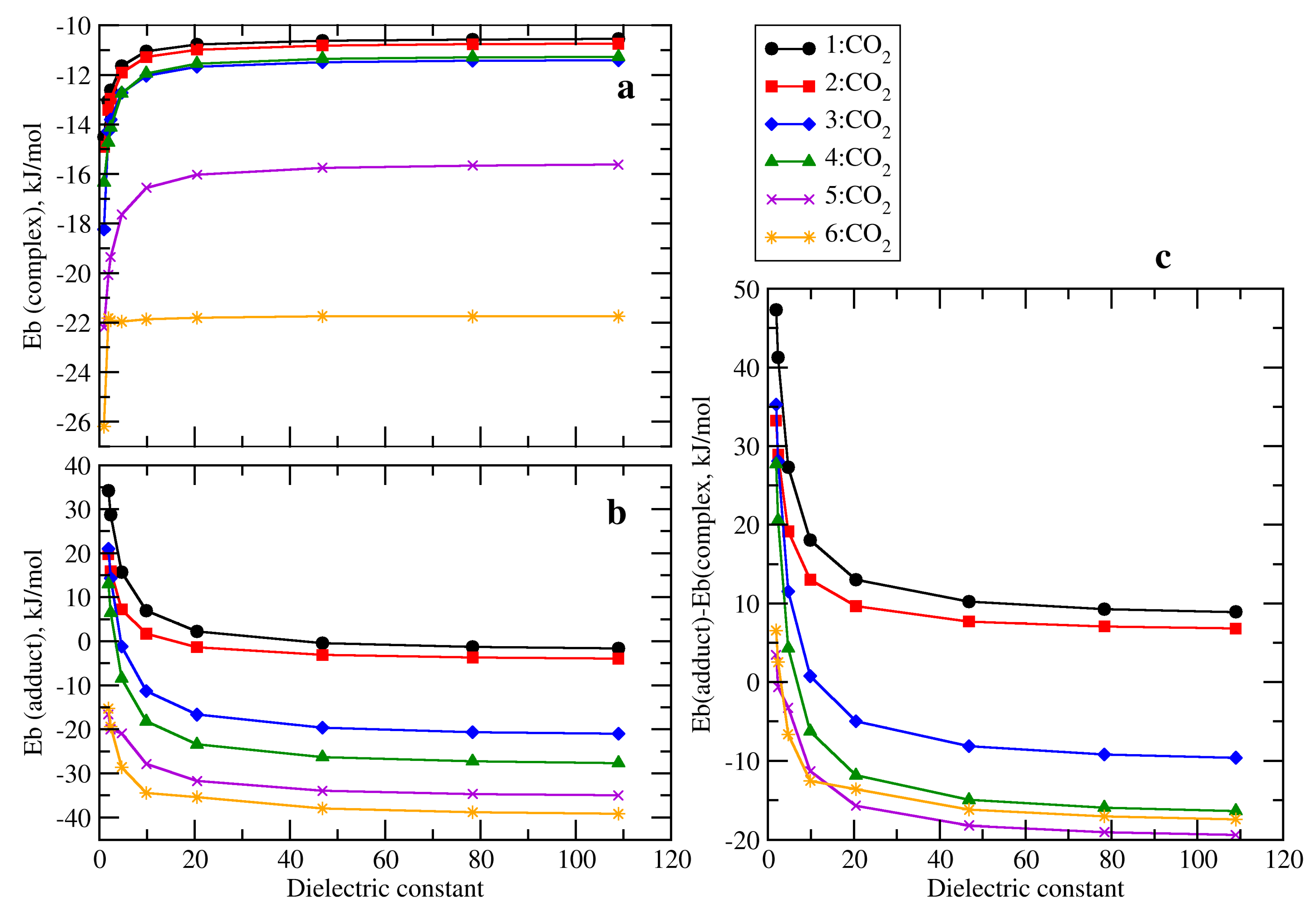
2.2. RR’R’’P + CO2 (2–5)
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alkorta, I.; Elguero, J. 1,2-Proton shifts in pyrazole and related systems: A computational study of [1,5]-sigmatropic migrations of hydrogen and related phenomena. J. Chem. Soc. Perkin Trans. 2 1998, 2497–2504. [Google Scholar] [CrossRef]
- Trujillo, C.; Sánchez-Sanz, G.; Alkorta, I.; Elguero, J. Computational Study of Proton Transfer in Tautomers of 3- and 5-Hydroxypyrazole Assisted by Water. Chem. Phys. Chem. 2015, 16, 2140–2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oziminski, W.P. The kinetics of water-assisted tautomeric 1,2-proton transfer in azoles: A computational approach. Struct. Chem. 2016, 27, 1845–1854. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Abboud, J.L.; Taft, R.W. The solvatochromic comparison method. 6. The π* scale of solvent polarities. J. Am. Chem. Soc. 1977, 99, 6027–6038. [Google Scholar] [CrossRef]
- Reichardt, C. Solvatochromic Dyes as Solvent Polarity Indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar] [CrossRef]
- Abraham, M.H. Hydrogen bonding. 31. Construction of a scale of solute effective or summation hydrogen-bond basicity. J. Phys. Org. Chem. 1993, 6, 660–684. [Google Scholar] [CrossRef]
- Catalán, J. Toward a Generalized Treatment of the Solvent Effect Based on Four Empirical Scales: Dipolarity (SdP, a New Scale), Polarizability (SP), Acidity (SA), and Basicity (SB) of the Medium. J. Phys. Chem. 2009, 113, 5951–5960. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Orozco, M.; Luque, F.J. Theoretical Methods for the Description of the Solvent Effect in Biomolecular Systems. Chem. Rev. 2000, 100, 4187–4226. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Li, J.; Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Density functional solvation model based on CM2 atomic charges. J. Phys. Chem. 1998, 109, 9117–9133. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C. Improvement of Anion Transport Systems by Modulation of Chalcogen Interactions: The influence of solvent. J. Phys. Chem. 2018, 122, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.-H.; Albrecht, M.; Raabe, G.; Pan, F.-F.; Räuber, C. Solvent-Dependent Enthalpic versus Entropic Anion Binding by Biaryl Substituted Quinoline Based Anion Receptors. J. Phys. Chem. 2015, 119, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Z.; Yuan, K.; Lv, L.-L.; Zhu, Y.-C.; Yuan, Z. Designation and Exploration of Halide–Anion Recognition Based on Cooperative Noncovalent Interactions Including Hydrogen Bonds and Anion−π. J. Phys. Chem. 2015, 119, 5842–5852. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, H.; Zhu, X.; Liu, H.; Zhu, W. Effects of solvent on weak halogen bonds: Density functional theory calculations. Int. J. Quantum Chem. 2011, 112, 1421–1430. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Crowe, D.; Nicholson, A.; Fleming, A.; Carey, E.; Kelleher, F. Conformational studies of Gram-negative bacterial quorum sensing acyl homoserine lactone (AHL) molecules: The importance of the n→ π* interaction. Biophys. Chem. 2018, 238, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Crowe, D.; Nicholson, A.; Fleming, A.; Carey, E.; Sánchez-Sanz, G.; Kelleher, F. Conformational studies of Gram-negative bacterial quorum sensing 3-oxo N-acyl homoserine lactone molecules. Biorg. Med. Chem. 2017, 25, 4285–4296. [Google Scholar] [CrossRef] [PubMed]
- Keeling, C.D.; Bacastrow, R.B. Energy and Climate: Studies in Geophysics. In Impact of Industrial Gases on Climate; The National Academies Press: Washington, DC, USA, 1977; pp. 72–95. [Google Scholar]
- Keeling, R.F.; Shertz, S.R. Seasonal and interannual variations in atmospheric oxygen and implications for the global carbon cycle. Nature 1992, 358, 723. [Google Scholar] [CrossRef]
- Keeling, C.D.; Whorf, T.P.; Wahlen, M.; van der Plichtt, J. Interannual extremes in the rate of rise of atmospheric carbon dioxide since 1980. Nature 1995, 375, 666. [Google Scholar] [CrossRef]
- Keeling, R.F.; Piper, S.C.; Heimann, M. Global and hemispheric CO2 sinks deduced from changes in atmospheric O2 concentration. Nature 1996, 381, 218. [Google Scholar] [CrossRef]
- Keeling, C.D.; Chin, J.F.S.; Whorf, T.P. Increased activity of northern vegetation inferred from atmospheric CO2 measurements. Nature 1996, 382, 146. [Google Scholar] [CrossRef]
- Keeling, R.F. Recording Earth’s Vital Signs. Science 2008, 319, 1771–1772. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.R.; Hawkins, E.; Jones, P.D. CO2, the greenhouse effect and global warming: From the pioneering work of Arrhenius and Callendar to today's Earth System Models. Endeavour 2016, 40, 178–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heard, D.E.; Saiz-Lopez, A. Atmospheric chemistry. Chem. Soc. Rev. 2012, 41, 6229–6230. [Google Scholar] [CrossRef] [PubMed]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef] [PubMed]
- Tomasko, D.L.; Li, H.; Liu, D.; Han, X.; Wingert, M.J.; Lee, L.J.; Koelling, K.W. A Review of CO2 Applications in the Processing of Polymers. Ind. Eng. Chem. Res. 2003, 42, 6431–6456. [Google Scholar] [CrossRef]
- Yang, H.; Xu, Z.; Fan, M.; Gupta, R.; Slimane, R.B.; Bland, A.E.; Wright, I. Progress in carbon dioxide separation and capture: A review. J. Environ. Sci. 2008, 20, 14–27. [Google Scholar] [CrossRef]
- Kanki, K.; Maki, H.; Mizuhata, M. Carbon dioxide absorption behavior of surface-modified lithium orthosilicate/potassium carbonate prepared by ball milling. Int. J. Hydrog. Energy 2016, 41, 18893–18899. [Google Scholar] [CrossRef]
- Tang, Z.; Lu, L.; Dai, Z.; Xie, W.; Shi, L.; Lu, X. CO2 Absorption in the Ionic Liquids Immobilized on Solid Surface by Molecular Dynamics Simulation. Langmuir 2017, 33, 11658–11669. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.W.; Mitchell, R.A.C. The effects of increasing CO2 on crop photosynthesis and productivity: A review of field studies. Plant Cell Environ. 1991, 14, 807–818. [Google Scholar] [CrossRef]
- Shukla, R.; Ranjith, P.; Haque, A.; Choi, X. A review of studies on CO2 sequestration and caprock integrity. Fuel 2010, 89, 2651–2664. [Google Scholar] [CrossRef]
- Chen, K.-H.; Tam, K.-W.; Chen, I.f.; Huang, S.K.; Tzeng, P.-C.; Wang, H.-J.; Chen, C. A systematic review of comparative studies of CO2 and erbium:YAG lasers in resurfacing facial rhytides (wrinkles). J. Cosmet. Laser Ther. 2017, 19, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lawal, A.; Stephenson, P.; Sidders, J.; Ramshaw, C. Post-combustion CO2 capture with chemical absorption: A state-of-the-art review. Chem. Eng. Res. Des. 2011, 89, 1609–1624. [Google Scholar] [CrossRef] [Green Version]
- Ingrosso, F.; Ruiz-López, M.F. Electronic Interactions in Iminophosphorane Superbase Complexes with Carbon Dioxide. J. Phys. Chem. A 2018, 122, 1764–1770. [Google Scholar] [CrossRef] [PubMed]
- Azofra, L.M.; Scheiner, S. Complexes containing CO2 and SO2. Mixed dimers, trimers and tetramers. Phys. Chem. Chem. Phys. 2014, 16, 5142–5149. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Blanco, F.; Elguero, J.; Dobado, J.A.; Ferrer, S.M.; Vidal, I. Carbon···Carbon Weak Interactions. J. Phys. Chem. A 2009, 113, 8387–8393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Carbenes as Electron-Pair Donors To CO2 for C···C Tetrel Bonds and C–C Covalent Bonds. J. Phys. Chem. A 2017, 121, 4039–4047. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Del Bene, J.E. Azines as Electron-Pair Donors to CO2 for N···C Tetrel Bonds. J. Phys. Chem. A 2017, 121, 8017–8025. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Carbon–Carbon Bonding between Nitrogen Heterocyclic Carbenes and CO2. J. Phys. Chem. A 2017, 121, 8136–8146. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Montero-Campillo, M.M.; Elguero, J. Trapping CO2 by Adduct Formation with Nitrogen Heterocyclic Carbenes (NHCs): A Theoretical Study. Chem. Eur. J. 2017, 23, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Montero-Campillo, M.M.; Alkorta, I.; Elguero, J. Binding indirect greenhouse gases OCS and CS2 by nitrogen heterocyclic carbenes (NHCs). PCCP 2018, 20, 19552–19559. [Google Scholar] [CrossRef] [PubMed]
- Pu, M.; Privalov, T. Ab Initio Molecular Dynamics with Explicit Solvent Reveals a Two-Step Pathway in the Frustrated Lewis Pair Reaction. Chem. Eur. J. 2015, 21, 17708–17720. [Google Scholar] [CrossRef] [PubMed]
- Denning, D.M.; Falvey, D.E. Solvent-Dependent Decarboxylation of 1,3-Dimethylimdazolium-2-Carboxylate. J. Org. Chem. 2014, 79, 4293–4299. [Google Scholar] [CrossRef] [PubMed]
- Denning, D.M.; Falvey, D.E. Substituent and Solvent Effects on the Stability of N-Heterocyclic Carbene Complexes with CO2. J. Org. Chem. 2017, 82, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- Torrent-Sucarrat, M.; Varandas, A.J.C. Carbon Dioxide Capture and Release by Anions with Solvent-Dependent Behaviour: A Theoretical Study. Chem. Eur. J. 2016, 22, 14056–14063. [Google Scholar] [CrossRef] [PubMed]
- Buß, F.; Mehlmann, P.; Mück-Lichtenfeld, C.; Bergander, K.; Dielmann, F. Reversible Carbon Dioxide Binding by Simple Lewis Base Adducts with Electron-Rich Phosphines. J. Am. Chem. Soc. 2016, 138, 1840–1843. [Google Scholar] [CrossRef] [PubMed]
- Mehlmann, P.; Mück-Lichtenfeld, C.; Tan, T.T.Y.; Dielmann, F. Tris(imidazolin-2-ylidenamino)phosphine: A Crystalline Phosphorus(III) Superbase That Splits Carbon Dioxide. Chem. Eur. J. 2017, 23, 5929–5933. [Google Scholar] [CrossRef] [PubMed]
- Tolman, C.A. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 1977, 77, 313–348. [Google Scholar] [CrossRef]
- Scheiner, S. A new noncovalent force: Comparison of P···N interaction with hydrogen and halogen bonds. J. Chem. Phys. 2011, 134, 094315. [Google Scholar] [CrossRef] [PubMed]
- Zahn, S.; Frank, R.; Hey-Hawkins, E.; Kirchner, B. Pnicogen Bonds: A New Molecular Linker? Chem. Eur. J. 2011, 17, 6034–6038. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. The Pnicogen Bond in Review: Structures, Binding Energies, Bonding Properties, and Spin-Spin Coupling Constants of Complexes Stabilized by Pnicogen Bonds. In Noncovalent Forces; Scheiner, S., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 191–263. [Google Scholar]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Electron density shift description of non-bonding intramolecular interactions. Comput. Theor. Chem. 2012, 991, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Legon, A.C. Tetrel, pnictogen and chalcogen bonds identified in the gas phase before they had names: A systematic look at non-covalent interactions. PCCP 2017, 19, 14884–14896. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Rozas, I.; Elguero, J. Molecular Complexes between Silicon Derivatives and Electron-Rich Groups. J. Phys. Chem. A 2001, 105, 743–749. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Barrios, L.; Rozas, I.; Elguero, J. Comparison of models to correlate electron density at the bond critical point and bond distance. J. Mol. Struc. THEOCHEM 2000, 496, 131–137. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H⋯F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Alkorta, I.; Solimannejad, M.; Provasi, P.; Elguero, J. Theoretical study of complexes and fluoride cation transfer between N2F+ and electron donors. J. Phys. Chem. A 2007, 111, 7154–7161. [Google Scholar] [CrossRef] [PubMed]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H. Gaussian-Basis Sets for Use in Correlated Molecular Calculations. 1. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Keith, T.A. AIMAll; 17.11.14B; TK Gristmill Software: Overland Park, KS, USA; Available online: http://aim.tkgristmill.com/ (accessed on 12 April 2018).
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D. Available online: http://www.jmol.org/ (accessed on 12 April 2018).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | 1:CO2 | 1/CO2 | 1-CO2 | ||||
|---|---|---|---|---|---|---|---|
| Eb | P···C | P···O | Eb | P···C | Eb | P···C | |
| Gas | −14.5 | 3.365 | 3.306 | ||||
| Hexane | −13.1 | 3.367 | 3.323 | 38.5 | 2.206 | 34.2 | 1.959 |
| Toluene | −12.6 | 3.372 | 3.332 | 36.0 | 2.245 | 28.7 | 1.946 |
| Chloroform | −11.6 | 3.387 | 3.349 | 31.6 | 2.318 | 15.7 | 1.926 |
| 1-Octanol | −11.1 | 3.397 | 3.362 | 29.5 | 2.358 | 7.0 | 1.917 |
| Acetone | −10.8 | 3.405 | 3.370 | 28.7 | 2.377 | 2.2 | 1.914 |
| Dimethylsulfoxide, DMSO | −10.6 | 3.409 | 3.374 | 28.2 | 2.387 | −0.4 | 1.912 |
| Water | −10.6 | 3.410 | 3.375 | 28.1 | 2.390 | −1.3 | 1.911 |
| Formamide | −10.6 | 3.410 | 3.374 | 28.1 | 2.391 | −1.6 | 1.911 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkorta, I.; Trujillo, C.; Sánchez-Sanz, G.; Elguero, J. Solvent and Substituent Effects on the Phosphine + CO2 Reaction. Inorganics 2018, 6, 110. https://doi.org/10.3390/inorganics6040110
Alkorta I, Trujillo C, Sánchez-Sanz G, Elguero J. Solvent and Substituent Effects on the Phosphine + CO2 Reaction. Inorganics. 2018; 6(4):110. https://doi.org/10.3390/inorganics6040110
Chicago/Turabian StyleAlkorta, Ibon, Cristina Trujillo, Goar Sánchez-Sanz, and José Elguero. 2018. "Solvent and Substituent Effects on the Phosphine + CO2 Reaction" Inorganics 6, no. 4: 110. https://doi.org/10.3390/inorganics6040110
APA StyleAlkorta, I., Trujillo, C., Sánchez-Sanz, G., & Elguero, J. (2018). Solvent and Substituent Effects on the Phosphine + CO2 Reaction. Inorganics, 6(4), 110. https://doi.org/10.3390/inorganics6040110