2.1. Halogen Oxoacids
Initially, we compared our computed geometries of the halogen oxoacids with experimental evidence. However, the experimental gas phase geometries are only available for all hypohalous acids and perchloric acid. For the rest of the HXO
n compounds, the comparison has been made with high-level ab initio calculations available in the literature. All of these geometrical data are gathered in
Table 1. The inspection of the results revealed that our calculated geometries are in good to excellent agreement with both the experimental and theoretical ones from the literature. As far as we are concerned, high-level ab initio calculations are not available for HIO
3, HBrO
4, and HIO
4. Therefore, this is the first time that such results are reported.
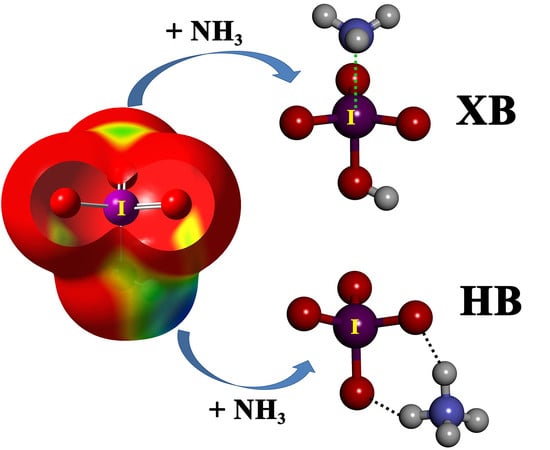
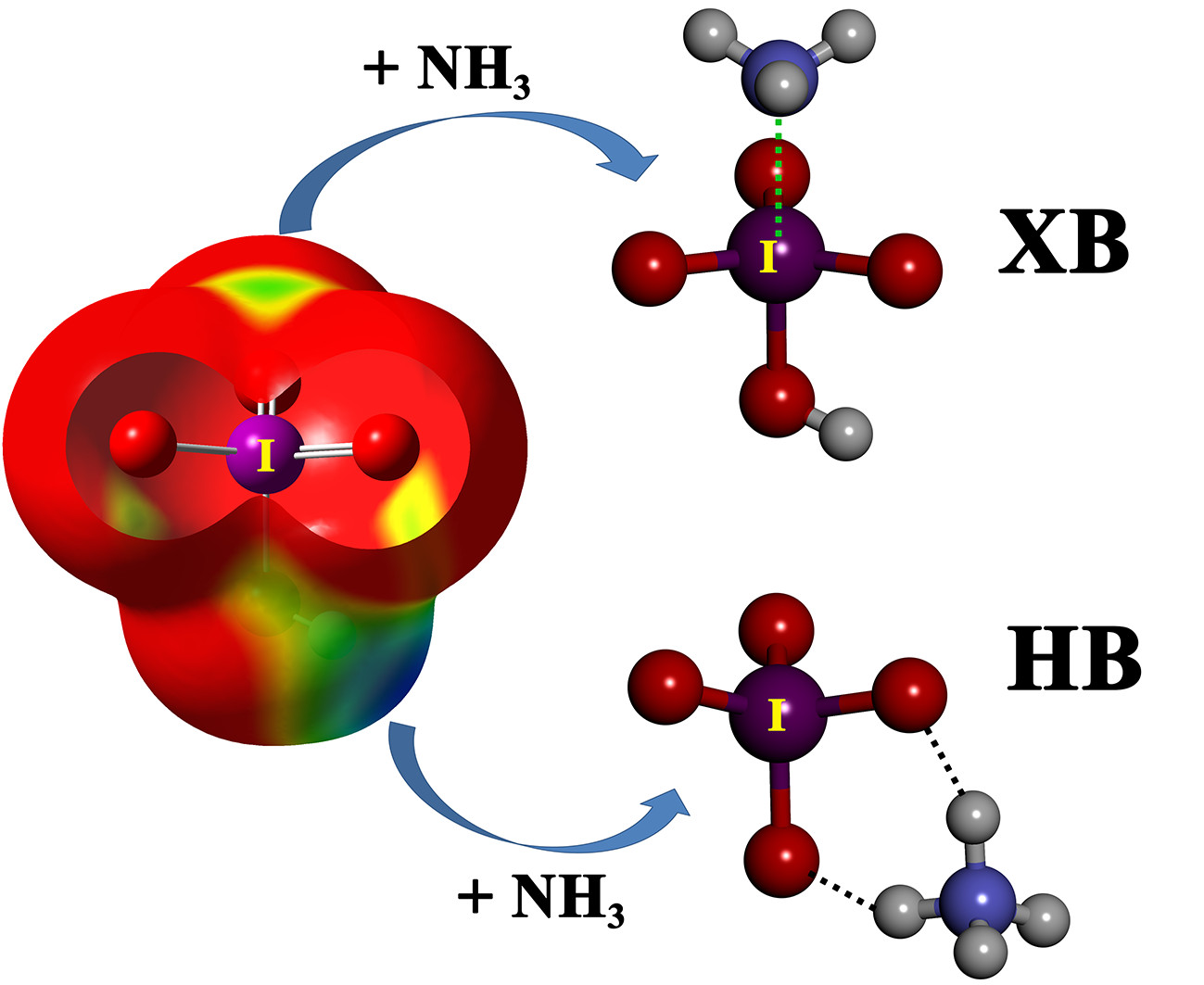
We have computed the molecular electrostatic potential (MEP) surfaces of the halogen oxoacids, paying special attention to the regions where the hydrogen and the halogen atoms are located, i.e., where the hydrogen and halogen bonding can be established (
Figure 1). The MEP values associated with these regions are gathered in
Table 2. A positive electrostatic potential region is located at the outermost region of both the H and halogen atoms in all cases, except for F in HFO, implying favorable formation of the complexes of HXO
n with electron-rich systems, such as NH
3 and Cl
−. In this regard, the MEP values for the hydrogen bonding interaction are always larger than the corresponding MEP values for the halogen bonding for the same oxoacid, suggesting that, from the electrostatic point of view, the HB complex will be the preferred arrangement. For example, the difference between HB and XB MEP values oscillates between 6.2 and 47.9 kcal·mol
−1 for HIO
3 and HClO
4, respectively. In the HB region, for the HXO and HXO
2 series of compounds, the MEP values diminish on going from X = F to X = I. The opposite is observed for the HXO
4 compounds, with MEP values increasing from X = Cl to X = I. However, for the HXO
3 oxoacids, the MEP values increase from X = Cl to X = Br to then decrease for X = I. In the XB region, the MEP values increase on going from X = Cl to X = I, as expected. For each X, the HB and XB MEP values follow different trends: the former values are very similar, except for HXO
4, which is sensibly larger, whereas the latter values increase from X = Cl to X = Br and decrease for X = I. In addition, we have compared the XB MEP values with the X–OH distances and found a good linear correlation (
R2 = 0.900,
Figure S1 of the
Supplementary Materials). However, no linear correlation is observed when comparing the O–H distances with the HB MEP values (
R2 = 0.248,
Figure S2 of the
Supplementary Materials). Thus the XB MEP results are able to reproduce the geometric results for the oxoacids, as opposed to the HB MEP results.
2.2. Geometrical Features and Energetics
We have considered two types of complexes between the halogen oxoacids and the Lewis bases, namely hydrogen-bonded (HB) complexes, formed through the interaction between either N atom (from ammonia) or Cl
− and the hydrogen atom of the oxoacid, and halogen-bonded (XB) complexes, formed through the interaction between N or Cl
− and the halogen atom of the oxoacid. At the RI-MP2/aug-cc-pVTZ level, both HB and XB complexes are local minima for all oxoacids, except for the fluorine derivative, for which the HB complex is the only one obtained, as expected from the abovementioned MEP results. In
Table 3 and
Table 4 we have put together the interaction energies and equilibrium distances for the NH
3 and Cl
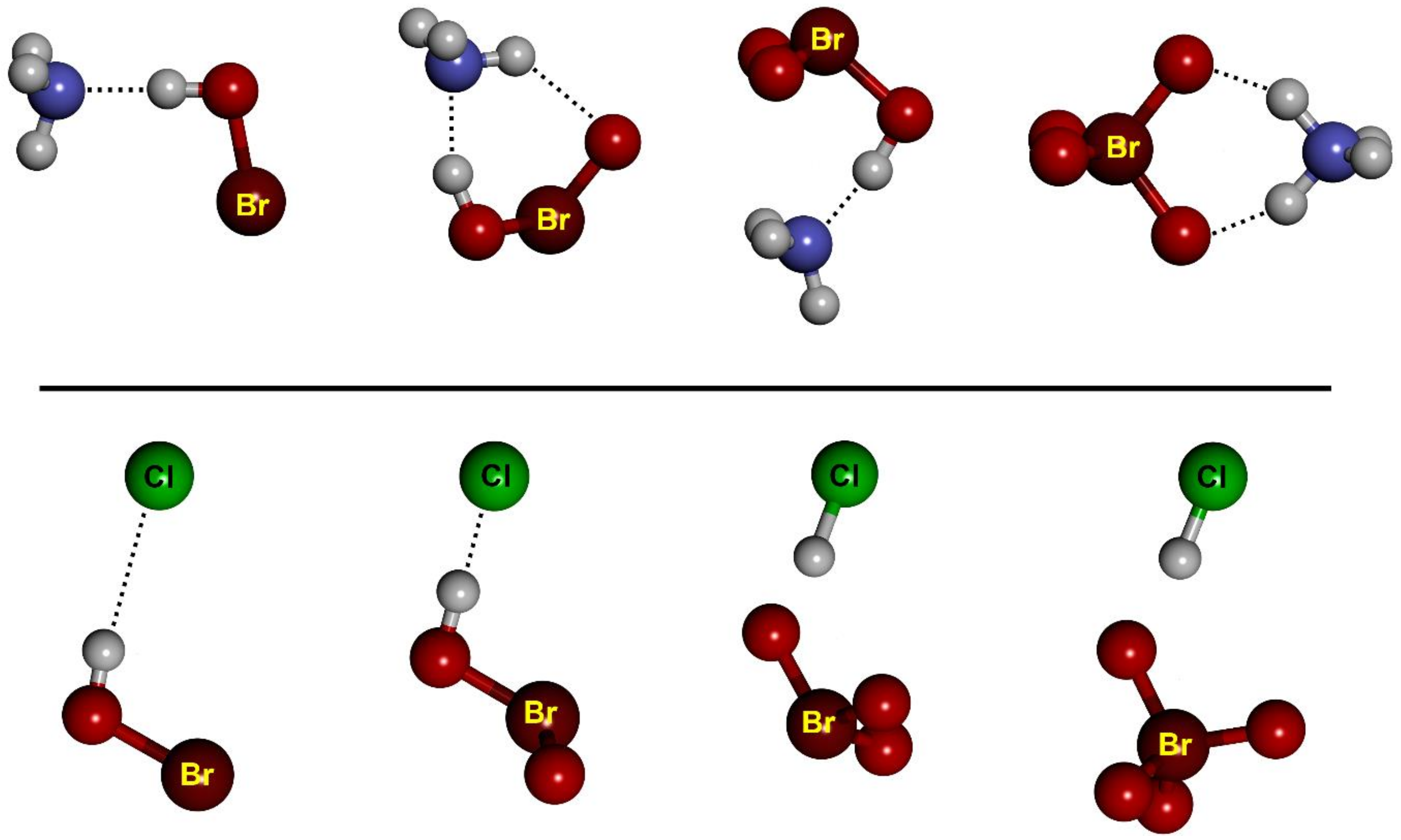
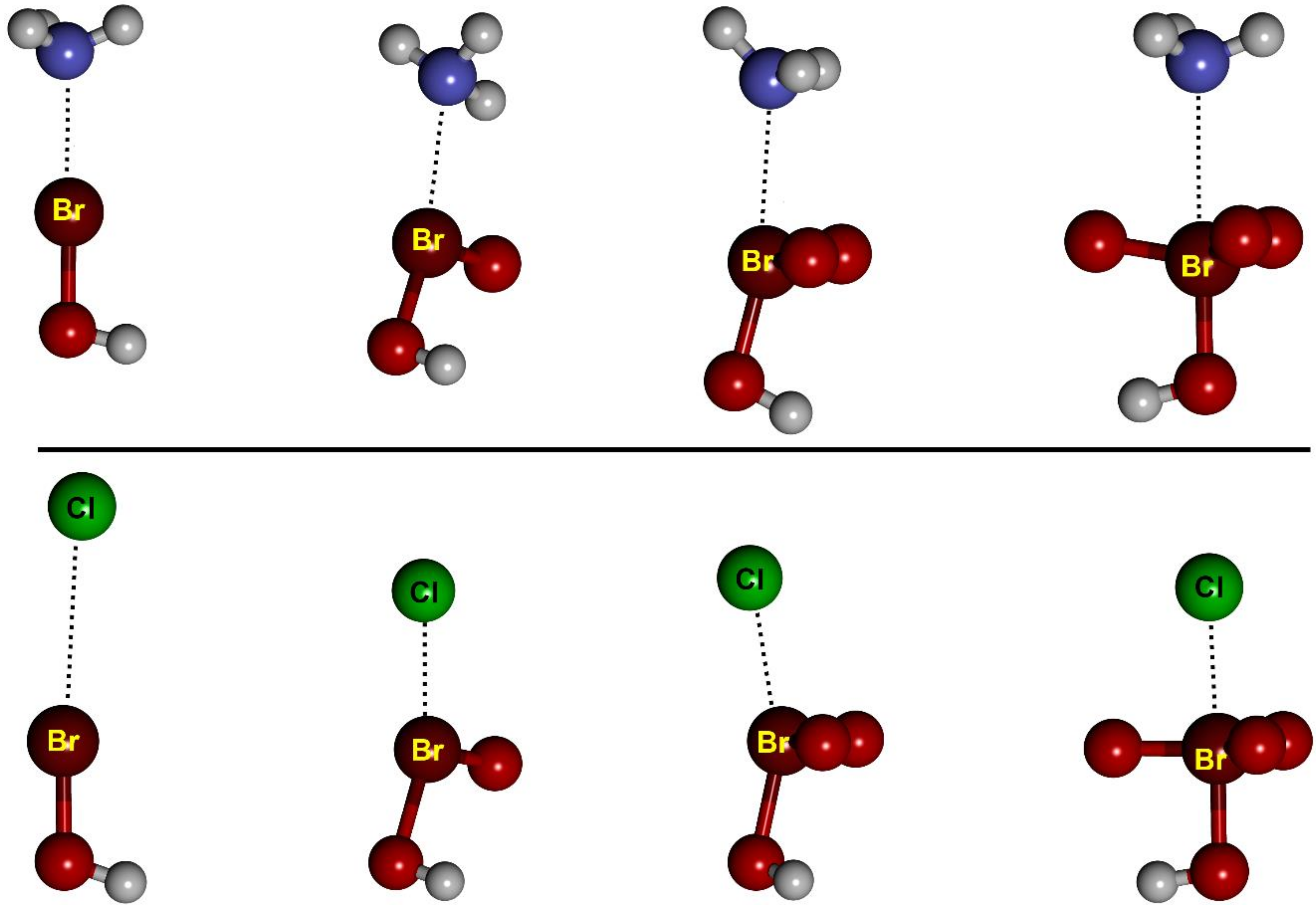
− complexes, respectively. The structure of the complexes is essentially independent of the nature of the halogen atom. For example, the geometry of all HB and XB HBrO
n complexes is reported in
Figure 2 and
Figure 3, respectively.
Let us first analyze the NH
3 HB complexes. From the inspection of the results (
Table 3), we observe that the N···H equilibrium distances range between 1.776 and 1.057 Å for HIO and HBrO
4 complexes, respectively. These distances are much shorter than the sum of the van der Waals radii of N and H atoms (2.86 Å) [
70]. These results indicate that the interaction established in these HB complexes is strong, as reflected in their interaction energies (Δ
E) that range between −10.7 and −20.8 kcal·mol
−1 for HBrO and HIO
4 complexes, respectively. For comparison purposes, the interaction energy of the H
3N···H
2O complex, computed at the MP2/aug-cc-pVTZ level, is −6.70 kcal·mol
−1 [
32], much smaller than our reported values. In fact, for some of them (HXO
4), a proton transfer is observed; the largest Δ
E values were observed for the HClO
4, HBrO
4, and HIO
4 complexes with NH
3 (−17.7, −20.1, and −20.8 kcal·mol
−1, respectively), which have quite short interatomic N···H distances (1.126, 1.057, and 1.061 Å, respectively) accompanied by the expected lengthening of the H–O distance by 0.429, 0.680, and 0.675 Å, respectively. The smallest Δ
E values and the largest intermolecular HB distances were obtained for the HXO:NH
3 complexes, and are constant for every halogen atom (ca. −11 kcal·mol
−1 and ca. 1.77 Å, respectively). Nearly constant Δ
E values were also observed for the HXO
2 complexes, though a little bit larger than for the HXO ones. In fact, the N···H distances are the same for both HXO and HXO
2 complexes. Particularly, the HXO
2 complexes show an additional intermolecular interaction, an HB between the X=O oxygen atom, and an NH
3 hydrogen atom, with increasing O···H distances (2.148, 2.191, and 2.259 Å for X = Cl, Br, and I, respectively) due to the concomitant lengthening of the X=O distance. Significant variations appear for the HXO
3:NH
3 complexes, yielding larger Δ
E values and shorter intermolecular HB distances than those for the HXO and HXO
2 complexes. Thus, the interaction energy increases, the N···H distances shorten, and the O–H distances lengthen with the number of oxygen atoms because of the increasing electron-withdrawing character of the XO
n moiety, making the H atom much more acidic. Moreover, for
n = 3 and
n = 4, the Δ
E values of the HXO
n complexes are not constant, unlike for
n = 1 and
n = 2, obtaining the largest values for X = I, most likely due to the increasing impact that the oxygen atoms have on the highly polarizable iodine atom.
The results for the chloride HB complexes (
Table 4) follow the same trends that have just been mentioned for the NH
3 HB complexes. Thus, a good linear correlation (
R2 = 0.888,
Figure S3 of the
Supplementary Materials) is found when comparing Δ
E values of NH
3 and Cl
− HB complexes. Notwithstanding, this correlation is improved (
R2 = 0.960) when an outlier is removed (HIO
3). In this regard, Δ
E values for HXO, on one hand, and for HXO
2:Cl
− complexes, on the other hand, are constant. In addition, the Cl···H equilibrium distances range between 1.918 and 1.333 Å for HFO and HBrO
4 complexes, respectively, larger than the ones corresponding to the HB NH
3 complexes, because of the larger size of the Cl atom. Again, these distances are much shorter than the sum of the van der Waals radii of Cl and H atoms (3.02 Å) [
70]. The interaction is much stronger (Δ
E = −24.0 to −49.1 kcal·mol
−1 for HBrO and HIO
4 complexes, respectively) than that found for the HB NH
3 complexes because of the anionic nature of the chloride. Now, proton transfer is not only observed for HXO
4 but for HXO
3 complexes as well. The interaction energy increases, the HB distances shorten (Cl···H distances from 1.333 to 1.491 Å) to values quite close to the RI-MP2/aug-cc-pVTZ computed distance in hydrogen chloride (1.275 Å), and the O–H distances lengthen (within 0.292–0.633 Å range) with the number of oxygen atoms as found in the HB NH
3 complexes.
Now, let us analyze the XB complexes, starting with the NH
3 ones. From the inspection of the results, gathered in
Table 3, we observe that the N···X equilibrium distances range from 3.240 to 2.340 Å for HClO
4 and HIO
4 complexes, respectively, which, at the same time, are the ones with the smallest and largest Δ
E values (−2.2 and −20.2 kcal·mol
−1, respectively). All of these distances are much shorter than the sum of the van der Waals radii of either Cl, Br, or I and N atoms (3.48, 3.52, and 3.70 Å, respectively) [
70]. These results indicate that the interaction in these XB complexes is attractive, as can be interpreted from their negative Δ
E values. Despite the size of the halogen atom increases on going from Cl to I, the shortest and largest XB equilibrium distances were observed for the HIO
n and HClO
n complexes, with the HBrO
n complexes being intermediate between both. Another intimately related general trend was observed in the binding energies of all these complexes: their value decreases as the size of the halogen increases, a fact that can be related to the deeper σ-hole observed for the iodine derivatives (
Figure 1), as predicted by our MEP calculations, which results in quite short N···I distances. Thus, in each series of the HXO
n complexes, the most and least stable complexes correspond to the iodine and chlorine derivatives, respectively, with the bromine complexes being intermediate between both. For example, in the HXO
2:NH
3 series, the Δ
E values are −4.4, −6.9, and −12.5 kcal·mol
−1, with N···X distances of 2.888, 2.755, and 2.649 Å for X = Cl, Br, and I, respectively. On the other hand, in each series of complexes for a given n, the interaction energy increases from
n = 1 to
n = 3, i.e., as the number of oxygen atoms augments. For instance, in the HClO
n:NH
3 series, the Δ
E values are −4.3, −4.4, and −4.8 kcal·mol
−1 for
n = 1, 2, and 3, respectively. However, for
n = 4, the interaction energy drops to −2.2 kcal·mol
−1. The same trend is also observed for the HBrO
n:NH
3 series. Unlike HClO
n and HBrO
n, the Δ
E values of the iodine oxoacids increase all the way from
n = 1 to
n = 4. These results can be explained in terms of the accessibility of the halogen atom by the Lewis base. In free HClO
4 and HBrO
4, the O–X–OH bond angles are 103.7° and 102.7°, and the X=O bond lengths are 1.42 and 1.58 Å, whereas in HIO
4 that bond angle is a little bit smaller (101.5°) and, most importantly, the I=O bond length is much longer (1.76 Å), moving the three oxo oxygen atoms a little bit further away, resulting in less repulsion with the electron-rich N atom of NH
3 when approaching the HIO
4 molecule. To some extent, these interaction energy results are in agreement with the MEP results (the largest Δ
E values are obtained for the molecules with the largest HB MEP values,
Table 2), meaning that electrostatics plays an important role in the stabilization of the complexes.
Just as in the HB complexes, the results for the chloride XB complexes (
Table 4) follow the same trends as the NH
3 XB complexes, with interaction energies that are substantially more negative with Cl
− than NH
3. As a matter of fact, the comparison of Δ
E values of NH
3 and Cl
− XB complexes yields a good linear correlation (
R2 = 0.953,
Figure S4 of the
Supplementary Materials). Therefore, in each series of the complexes, the most and least stable complexes in the HXO
n series correspond to the iodine and chlorine derivatives, respectively. In addition, in each series of complexes for a given
n, the interaction energy increases from
n = 1 to
n = 3, and then drops for
n = 4, except for HIO
n for which the Δ
E values increase from
n = 1 to
n = 4. The Cl···X equilibrium distances range between 3.313 and 2.476 Å for HClO
4 and HBrO
4 complexes, respectively. As expected from the large and negative Δ
E values, these distances are much shorter than the sum of the van der Waals radii of either Cl, Br, or I and Cl atoms (3.64, 3.68, and 3.86 Å, respectively) [
70].
Comparison of the energetics of the complexes indicates that the only XB complexes that are more favored than the HB ones are HIO:NH3, HIO:Cl−, HIO2:Cl−, and HXO3:Cl−. Therefore, as the number of the O atoms increases, the XB complexes with an anionic Lewis base are being more favored than the HB ones, especially those with X = I (the halogen atom with the deepest σ-hole), except for those formed with HXO4 due to the accessibility of the X atom by the Lewis base.
2.3. Electron Density Analysis
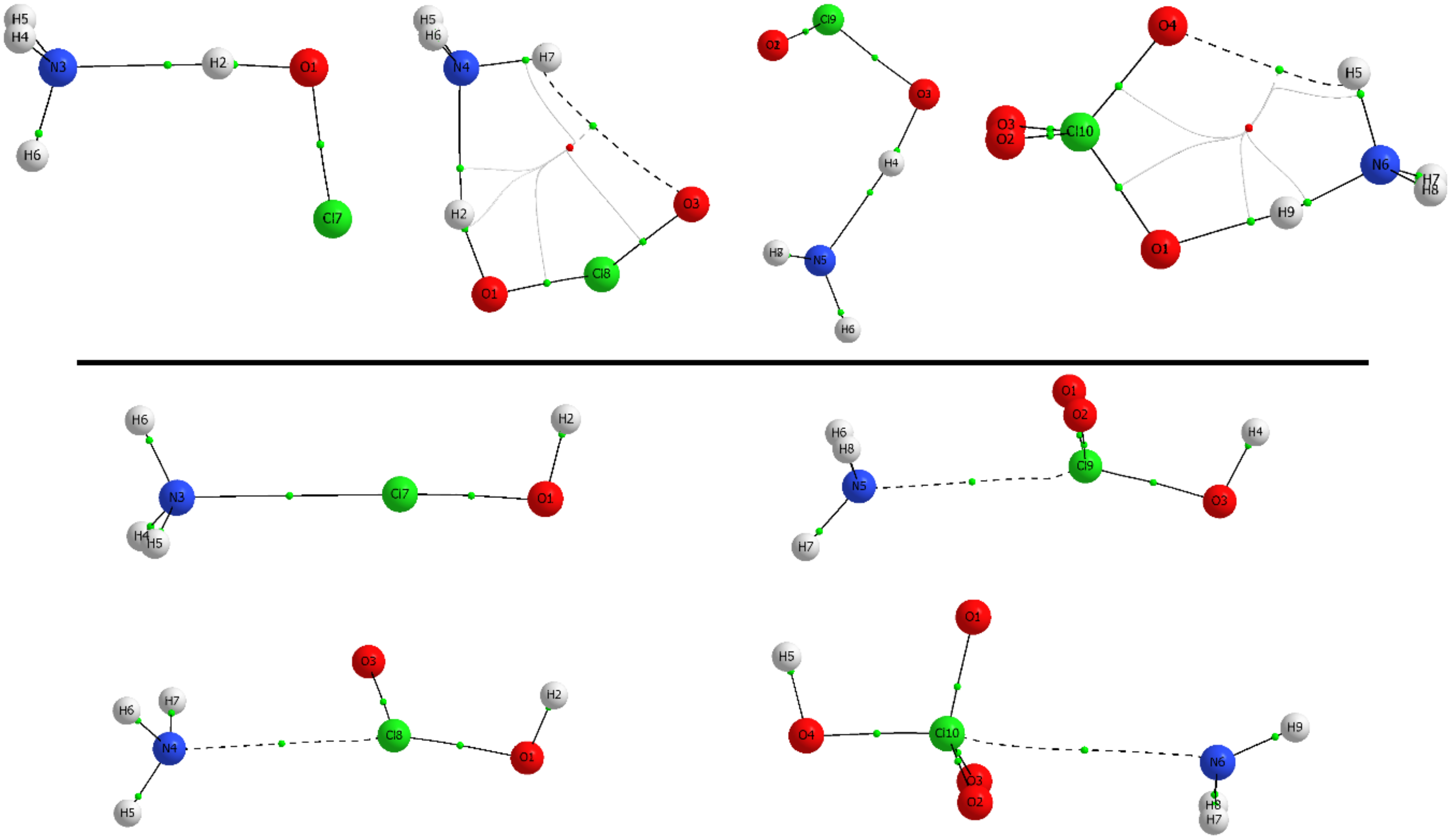
The AIM methodology was applied to evaluate the properties of the charge density for our systems. The topological analysis of the electron density reveals the presence of a single intermolecular bond critical point (BCP) that connects the interacting atom of the Lewis base (N or Cl) with either the H or X atom of HXO
n in HB and XB complexes, connected through the corresponding bond paths. For example, the molecular graphs of all HClO
n:NH
3 complexes are shown in
Figure 4. Moreover, for HXO
2:NH
3 and HXO
4:NH
3 HB complexes, an additional BCP is located between an H atom of NH
3 and an O atom of the oxoacid, giving rise to the formation of a ring critical point. At the intermolecular BCPs, the values of the electron densities,
ρ, range between 0.299 and 0.047 a.u. for HXO
n:NH
3 HB complexes, between 0.218 and 0.050 a.u. for HXO
n:Cl
− HB complexes, between 0.076 and 0.008 a.u. for HXO
n:NH
3 XB complexes, and between 0.084 and 0.011 a.u. for HXO
n:Cl
− XB complexes (
Table 5 and
Table 6). Moreover, very large and negative values of the Laplacian, ∇
2ρ, are obtained for the HB complexes where proton abstraction occurs (
Table 5), namely all HXO
4 HB complexes and HXO
3:Cl
− HB ones, consistent with the existence of a covalent bond. For the rest of the HB complexes, HXO
n:Cl
− XB complexes (except that with HClO
4), HIO
n:NH
3 complexes, and the HBrO:NH
3 complex, positive ∇
2ρ values are accompanied by negative values of the total electron density energy, (
Table 5), which indicate a partial covalent nature of these interactions [
71,
72]. For the rest of the XB complexes, the positive values accompanying ∇
2ρ and
H, in addition to the low
ρ values, are genuine descriptors of charge-depleted intermolecular regions at BCPs, expected of closed-shell interactions [
73,
74,
75,
76,
77,
78,
79,
80,
81].
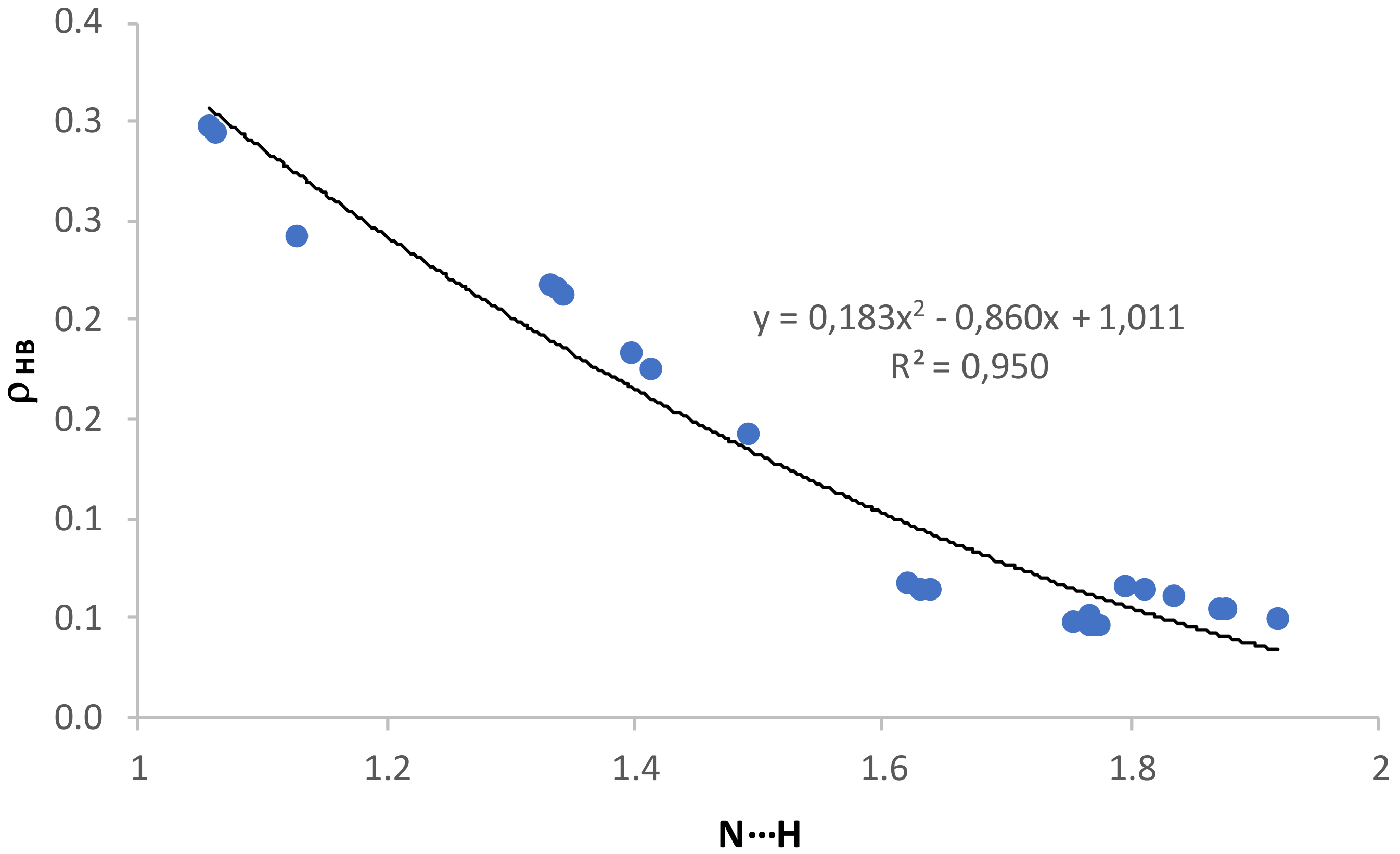
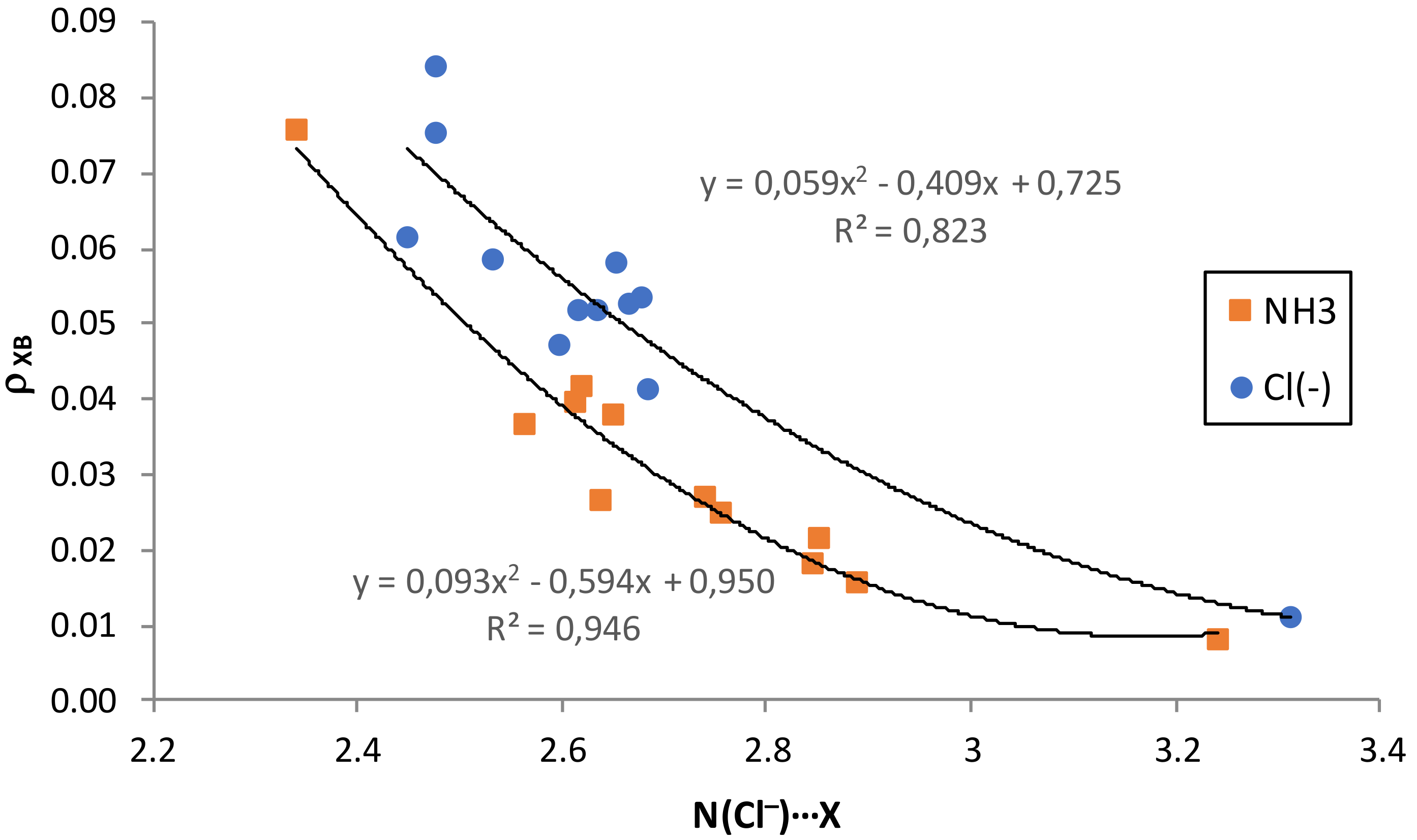
The graphical representations of the electron density at the intermolecular BCP versus the intermolecular equilibrium distance (
Figure 5) have good second-order polynomial trendlines, with correlation coefficients
R2 = 0.950 for all HB complexes, and 0.946 and 0.823 for NH
3 and Cl
− XB complexes, respectively. There is also an excellent second-order polynomial fitting for the representation of
ρ at the intermolecular BCP in HB complexes,
ρHB, versus the O–H distance (
R2 = 0.999 and 1.0 for NH
3 and Cl
− complexes, respectively,
Figure S5 of the
Supplementary Materials).
A comparison of the binding energies with the properties of the intermolecular BCPs reveals an interesting trend: the higher the electron density at the BCP, the stronger the interaction. The same trend applies to the Laplacian. In fact, there is a very good polynomial correlation (
Figures S6 and S7 of the
Supplementary Materials) between the interaction energies and the values of either ∇
2ρHB (
R2 = 0.837–0.975) or
ρXB (
R2 = 0.824–0.995). These results point out that the
ρXB and ∇
2ρHB values can be used as a measure of bond order for this type of interaction. This is in line with other studies, especially on hydrogen-bonded systems, where it was found that the
ρ value is a good descriptor of the strength of the interaction [
75,
82].
2.4. Natural Bond Orbital Analysis
A Natural Bond Orbital (NBO) analysis was carried out to further analyze the geometrical, energetical, and topological characteristics of these HB and XB interactions between the halogen oxoacid derivatives and the Lewis bases. We have examined all possible intermolecular interactions between occupied (donor) Lewis-type NBOs and vacant (acceptor) non-Lewis NBOs and estimated their energetic importance by the second-order perturbation theory. According to the NBO analysis, the interaction is primarily based on a charge donation from the lone pairs of either the N atom of NH
3 or chloride Cl to the vacant σ* antibonding orbital of the H–O bond in the HB complexes of HXO
n [Lp(N) → σ*(H–O)], and to the vacant σ* antibonding orbital of the X–OH bond in the HXO
n XB complexes, as derived from the calculated second-order orbital perturbation energies (
E(2)) listed in
Table 7. In the case of the HB complexes, very large
E(2)HB values were obtained (between 56.8 and 29.2 kcal·mol
−1 for the NH
3 complexes and between 80.8 and 49.8 kcal·mol
−1 for the Cl
− ones) due to the proximity of the interacting groups. Significant
E(2)XB values were also obtained for the XB complexes, with increasing values as we move from X = Cl to X = I. Thus,
E(2)XB is maximum for HIO
n complexes, the ones with the deepest σ-holes, and minimum for HClO
n complexes, the ones with the shallowest σ-holes. In fact, in nine of the HB complexes (where a proton transfer was observed) and two of the XB complexes, the NBO method is not able to properly recognize the constituent molecules (
Table 7) most likely due to the proximity of the interacting groups. Consequently, a significant charge is transferred from either the NH
3 or Cl
− unit to the oxoacid, even in the cases when no proton transfer complexes are considered (up to 0.105
e and 0.181
e in NH
3 and Cl
− HB complexes, and up to 0.179
e and 0.437
e in NH
3 and Cl
− XB complexes). In the case of the XB complexes, the
E(2) values for the NH
3 complexes (between 28.4 and 0.3 kcal·mol
−1) are smaller than the corresponding ones for their HB counterparts. This is not the case for Cl
− XB complexes, with
E(2)XB values that are larger than their HB counterparts for HXO complexes.
The secondary intermolecular interaction found by means of the AIM analysis in HXO2:NH3 HB complexes is also found in the NBO analysis [Lp(O) → σ*(H–N)], though they contribute with very small E(2) values (0.7, 1.0, and 1.5 kcal·mol−1 for X = Cl, Br, and I, respectively) compared to the ones that were obtained for the primary HB interaction.
In addition, we have also compared the charge transfer with the second-order orbital perturbation energies, obtaining a very good linear correlation for NH
3 and Cl
− HB (
R2 = 0.995 and 0.951, respectively,
Figure S8 of the
Supplementary Materials) and XB (
R2 = 0.978 (excluding HIO
4) and 0.955, respectively,
Figure S9 of the
Supplementary Materials) complexes. Therefore, the larger the
E(2) energy, the larger the charge transfer, irrespective of the nature of the complexes.
2.5. Energy Partition Scheme
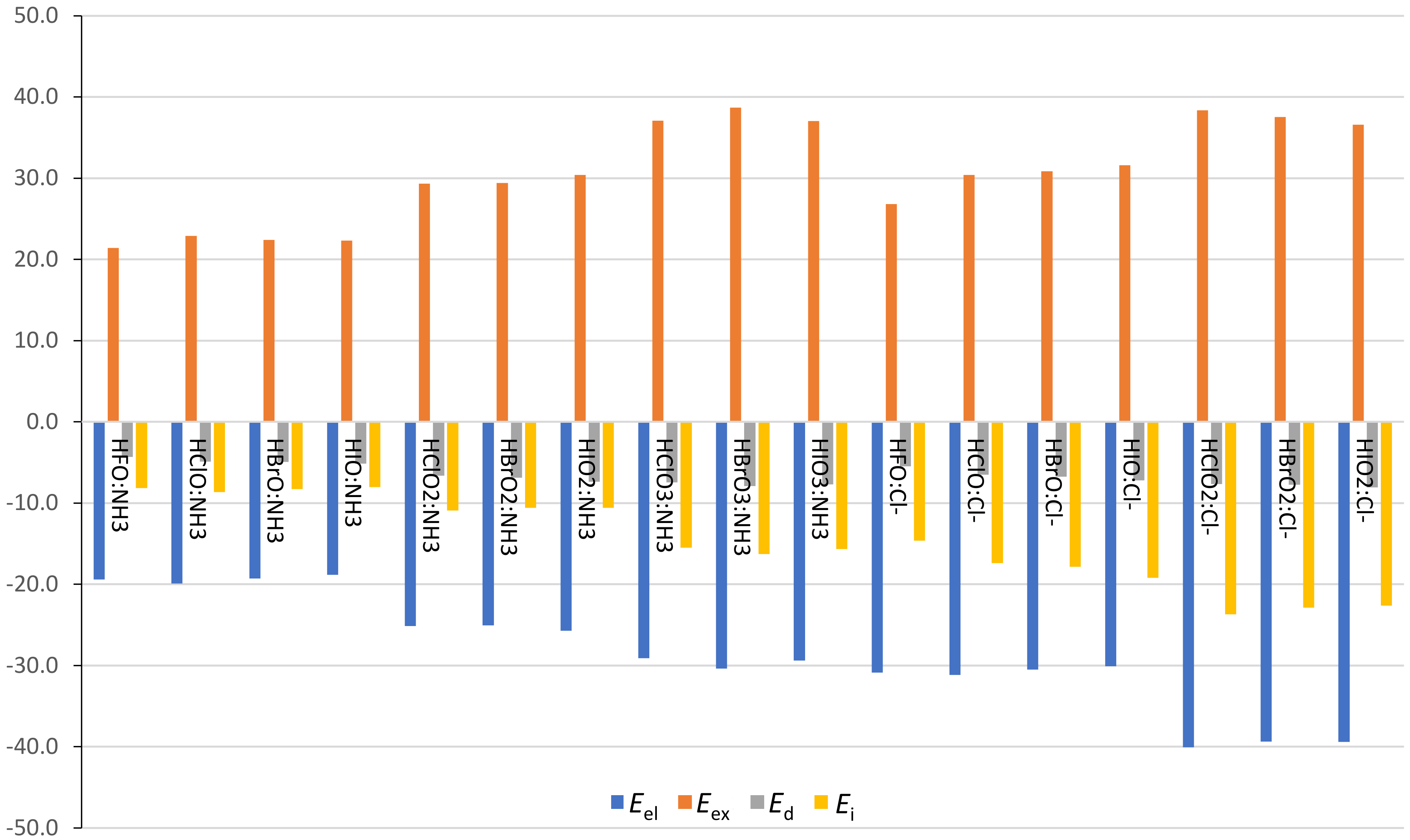
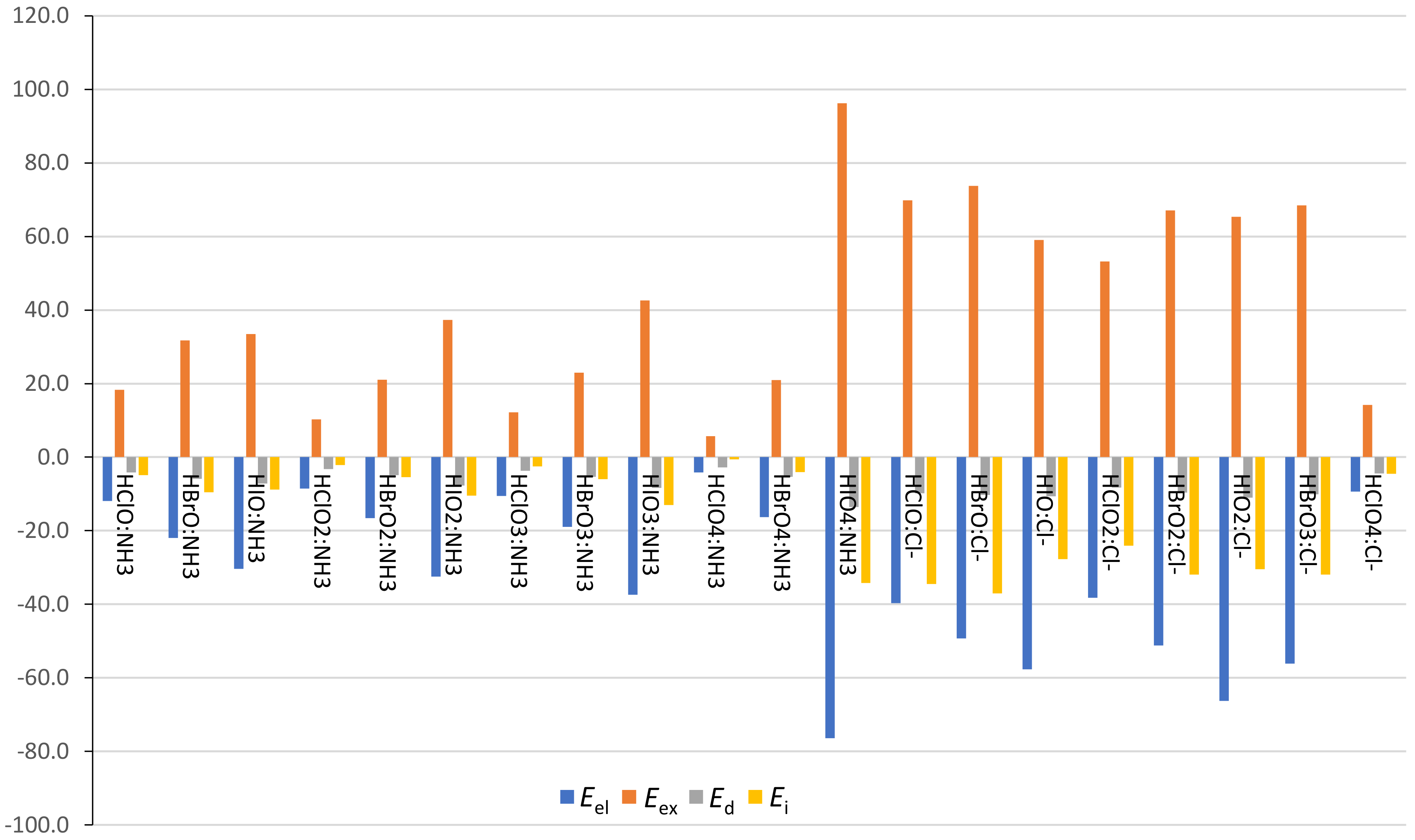
The physical nature of these HB and XB interactions has been analyzed by means of SAPT calculations. The energy contributions obtained from the SAPT partitioning scheme are listed in
Table S1 (HB) and
Table S2 (XB) of the
Supplementary Materials and the corresponding terms are represented in
Figure 6 (HB) and
Figure 7 (XB). Some complexes have been removed from the analysis (proton transfer complexes and HClO
3, HBrO
4, and HIO
4 XB complexes with Cl
−) because they yield misleading results, most likely due to the proximity of the interacting units. The SAPT analysis revealed that the electrostatic term,
Eel, is negative for all complexes, larger for anionic than for neutral complexes, with values ranging from −34.0 to −18.8 kcal·mol
−1 and from −40.1 to −30.5 kcal·mol
−1 for HXO
n:NH
3 and HXO
n:Cl
− HB complexes, respectively. The same behavior is observed for XB complexes, with
Eel values between −76.4 and −4.2 kcal·mol
−1 and between −77.3 and −9.4 kcal·mol
−1 for HXO
n:NH
3 and HXO
n:Cl
− complexes, respectively.
Eel is the most important attractive contribution in all cases, with a relative weight within all attractive forces greater than 53.2% in HB complexes and greater than 47.3% in XB complexes. In addition, the electrostatic contribution becomes larger with the increasing number of O atoms in HB complexes. For instance, the Eel values are −19.9, −25.1, and −29.1 kcal·mol−1 for HClO:NH3, HClO2:NH3, and HClO3:NH3 HB complexes, respectively. In XB complexes, however, the Eel values increase on going from X = Cl to X = I, due to the smallest and largest σ-holes for these atoms, respectively, as observed in HXO:NH3 XB complexes (Eel = −11.9, −22.0, and −30.4 kcal·mol−1 for X = Cl, Br, and I, respectively). In general, the induction term, Ei, is the second most important attractive contribution. The Eel term, Ei, becomes larger with the number of oxygen atoms in HB complexes, as it usually increases with the size of the X atom in XB complexes. The smallest contribution, the dispersion energy, Ed, is kept more or less constant within each HXOn HB series and, at the same time, it becomes larger within each HXOn XB series, as expected from the high polarizability of the heavier X atoms.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}