
Using Real Time Measurements to Derive the Indoor and Outdoor Contributions of Submicron Particulate Species and Trace Gases




Abstract
:1. Introduction
2. Materials and Methods
2.1. Measurement Facility
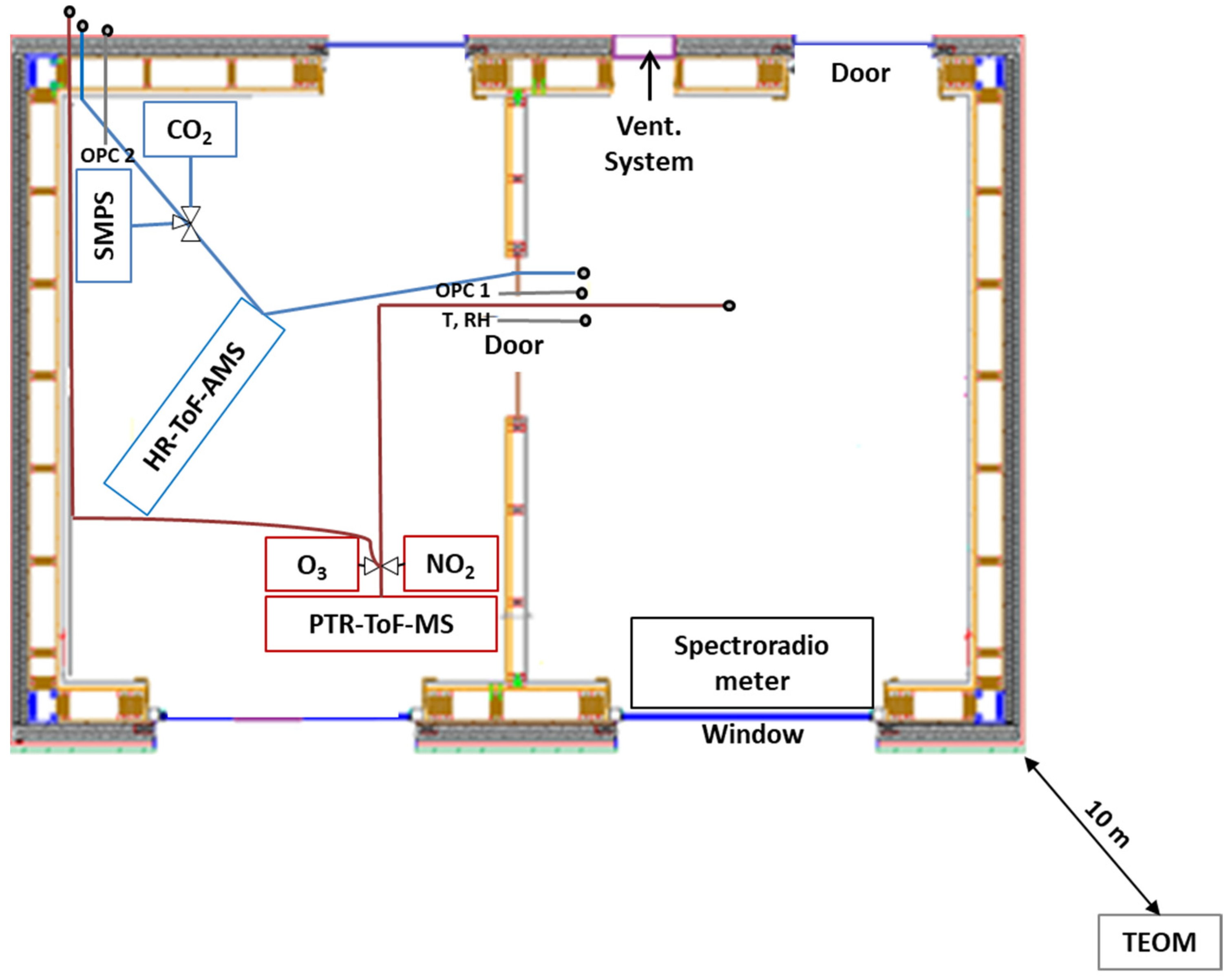
2.2. Sampling Setup, Instrumentation and Measurement Procedures
2.3. Data Treatment for Online Mass Spectrometers
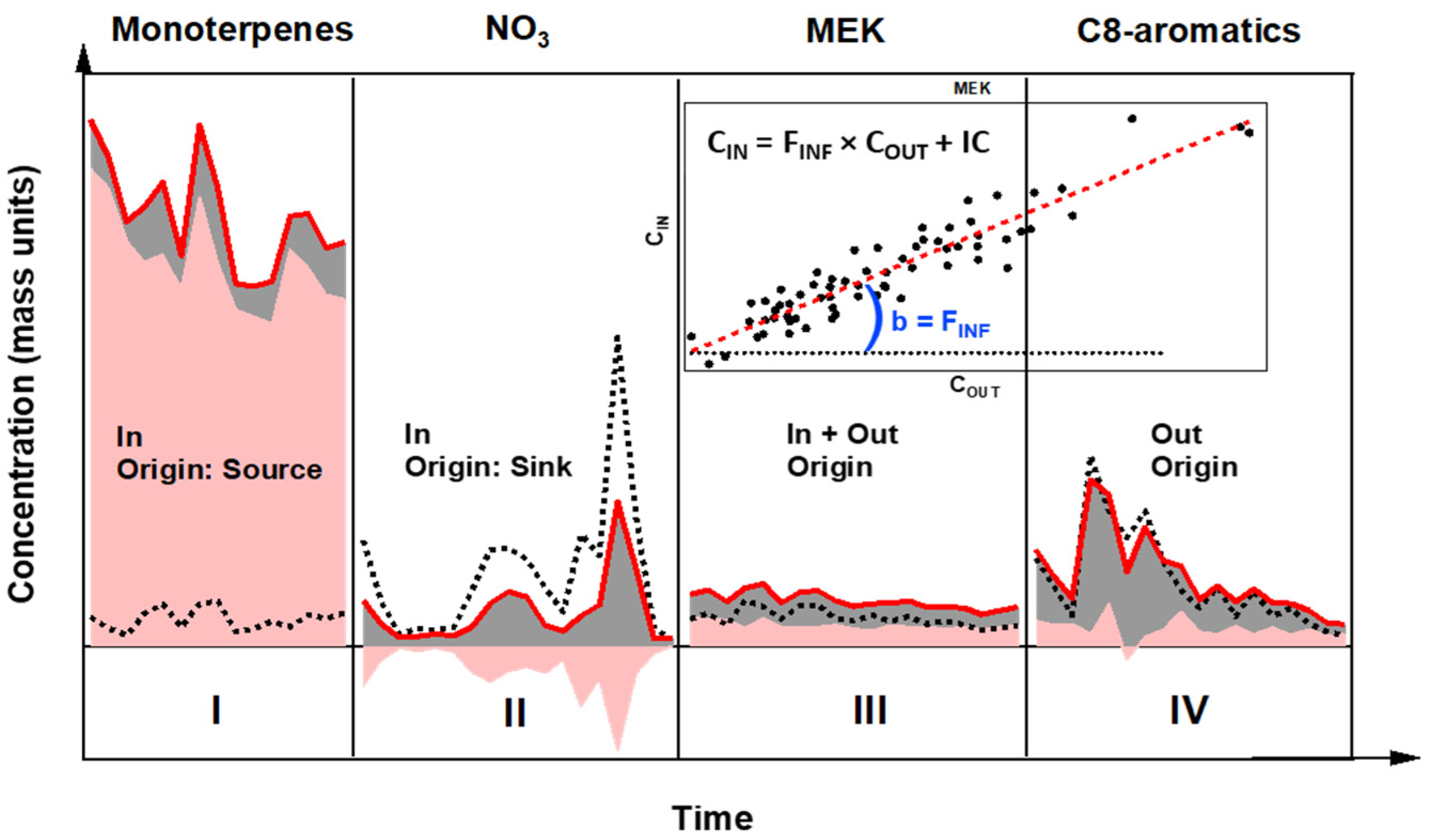
2.4. Indoor/Outdoor Apportionment Methodology
3. Results
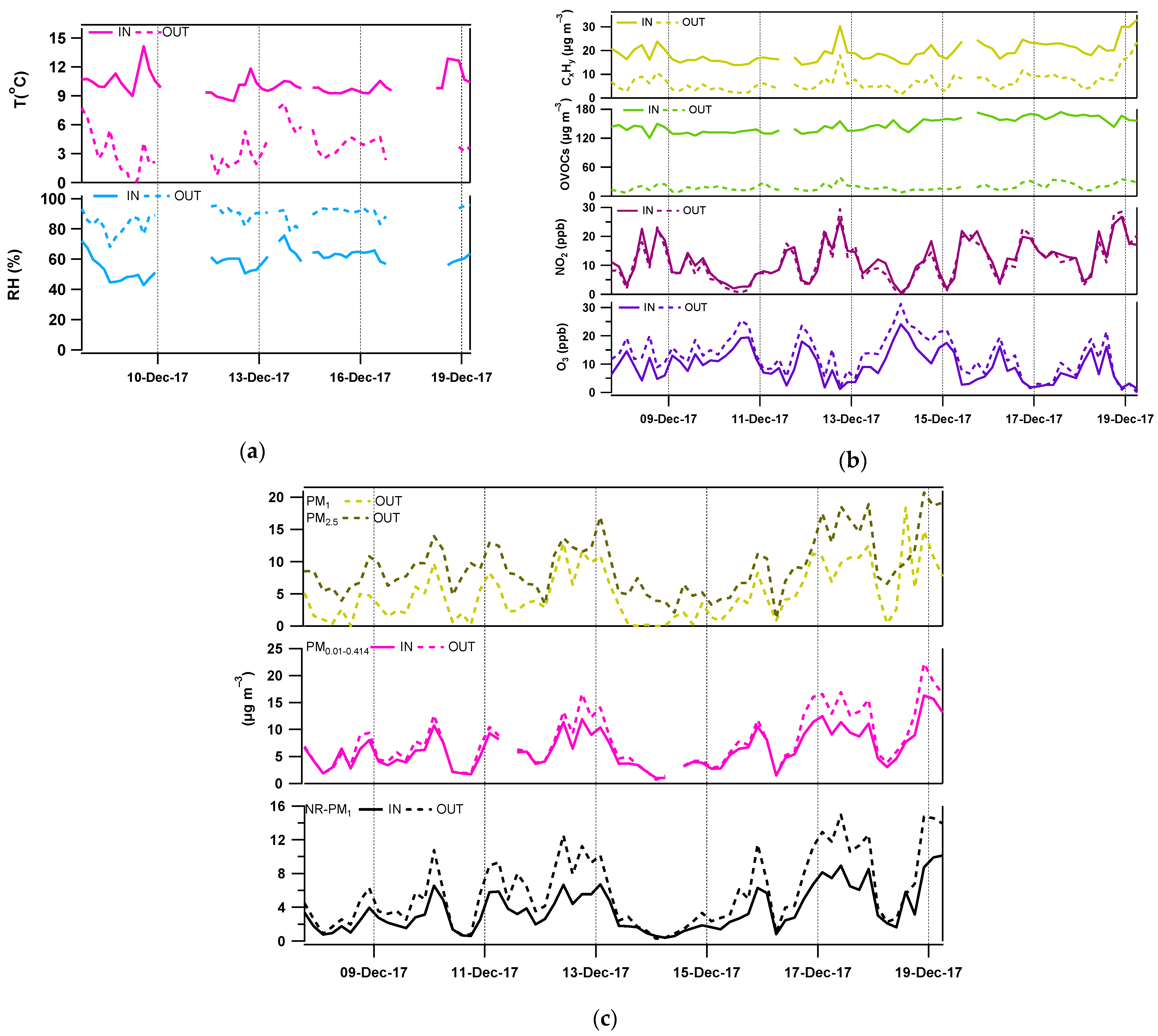
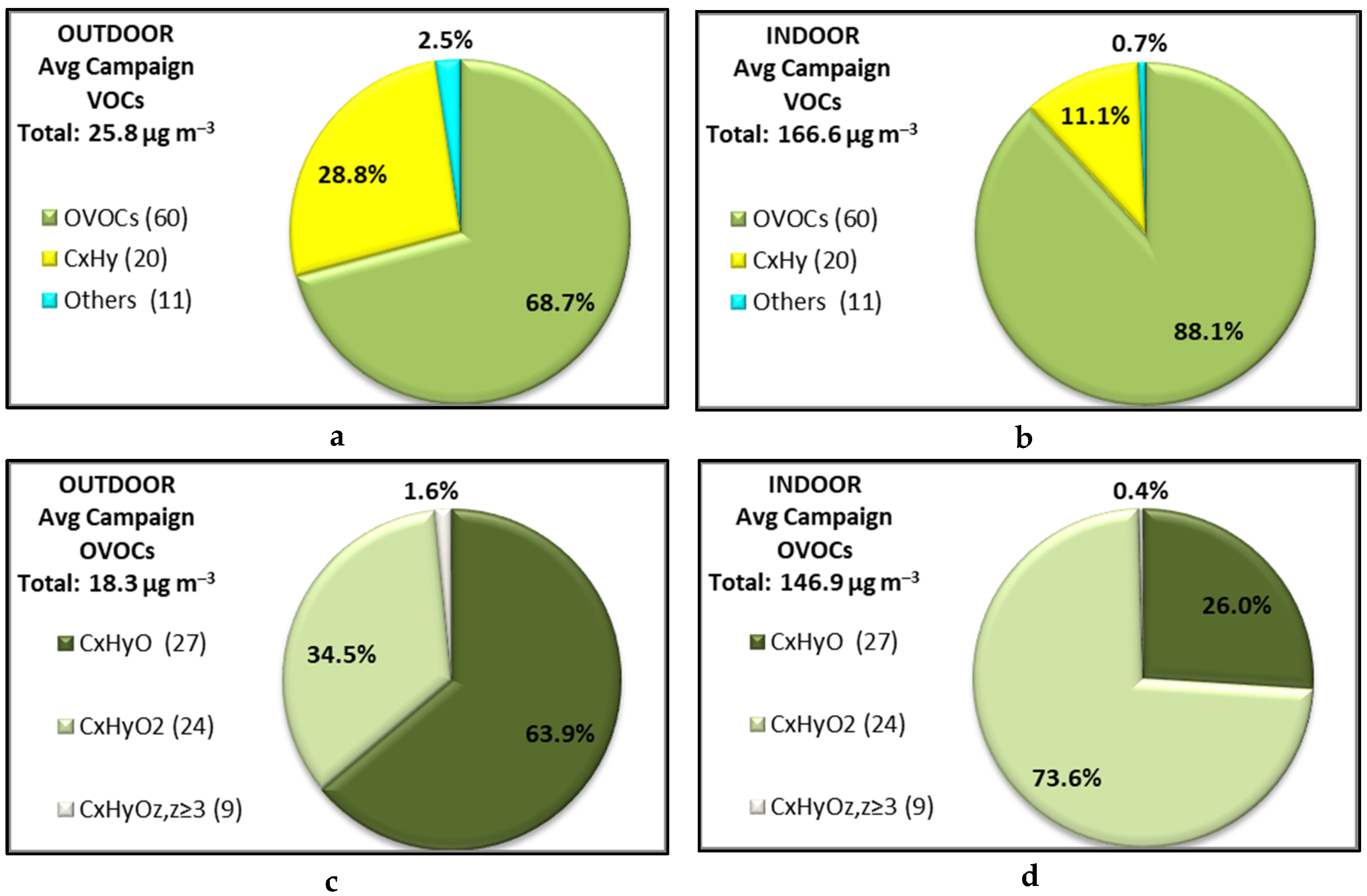
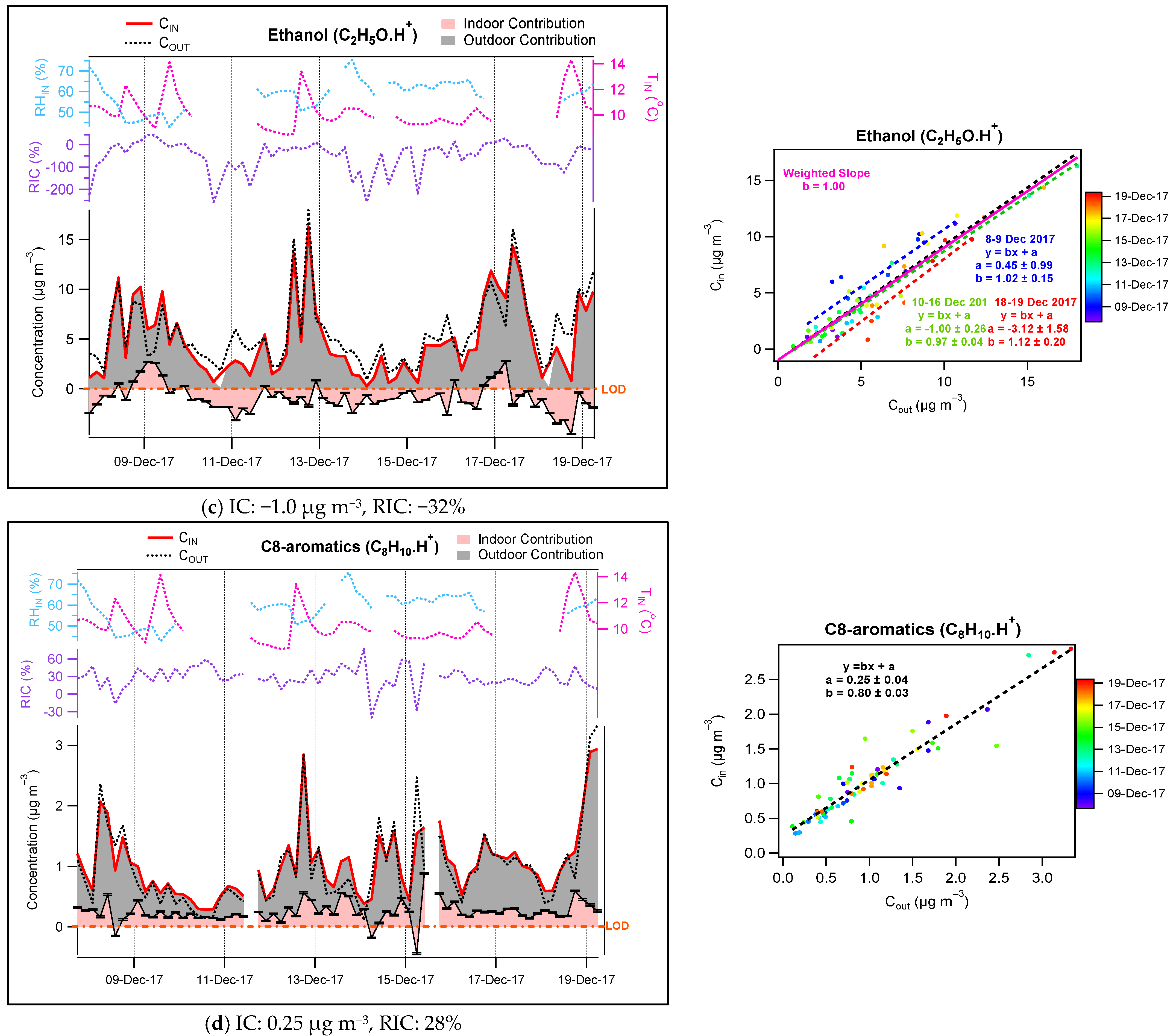
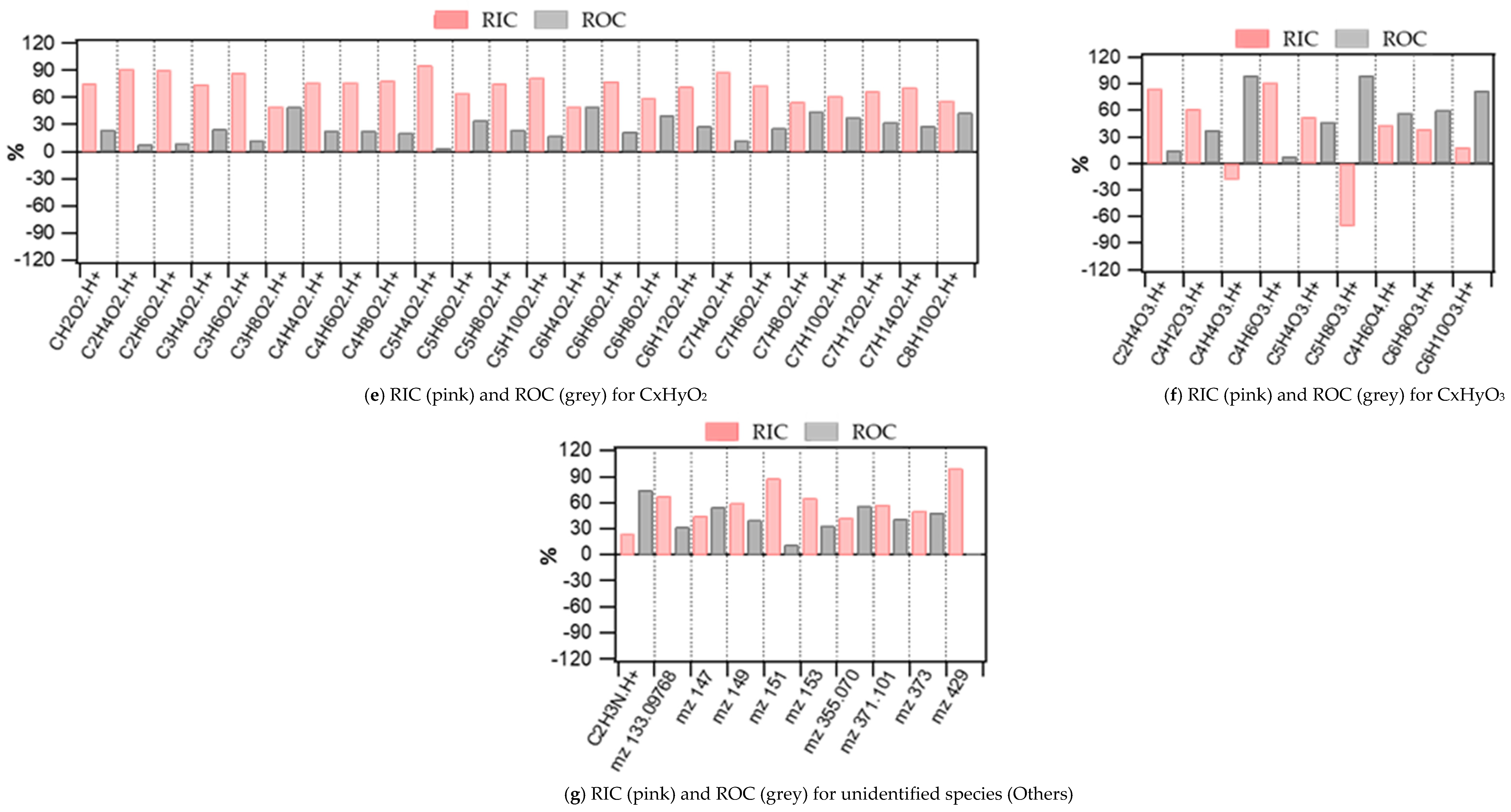
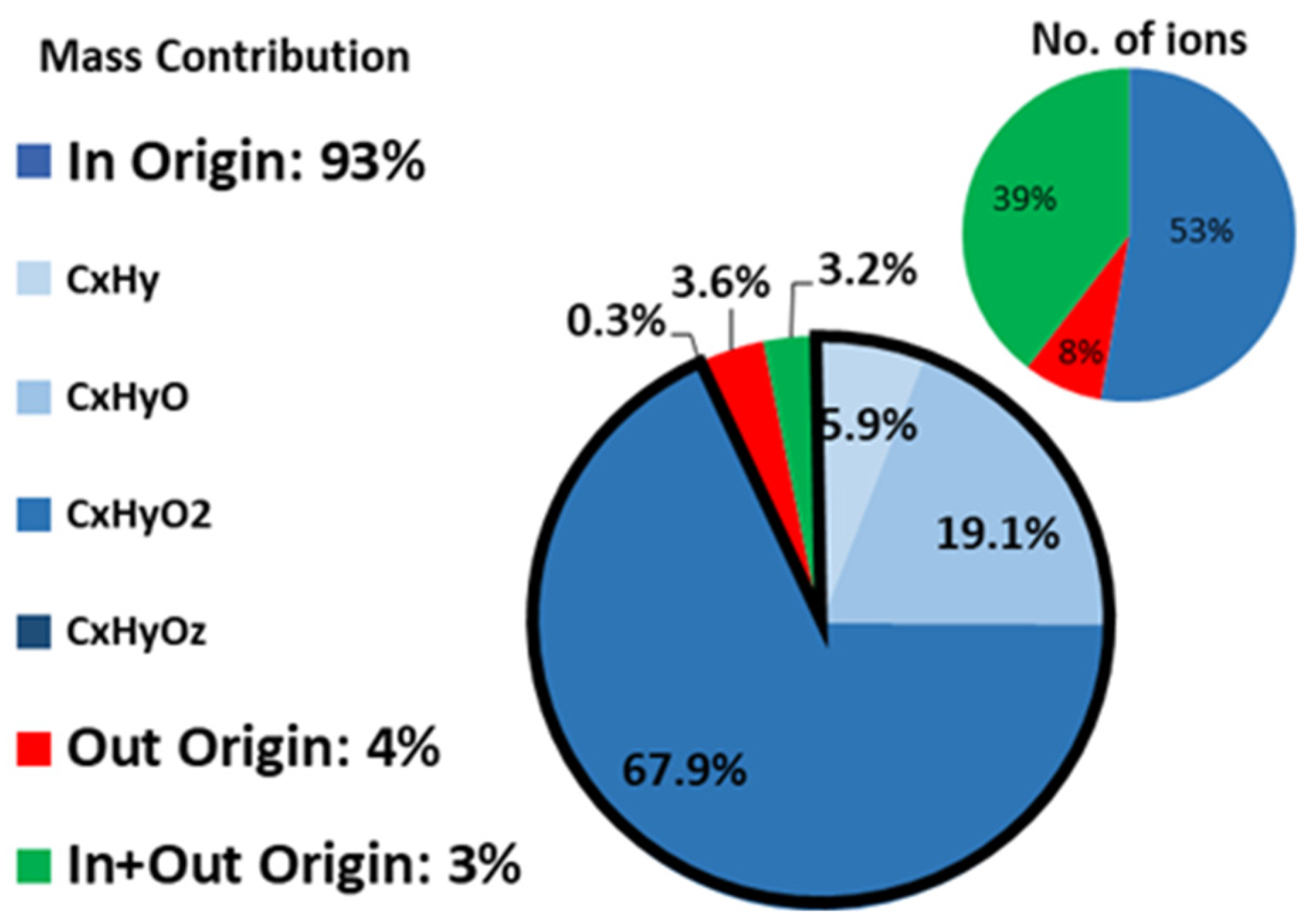
3.1. Indoor and Outdoor Composition
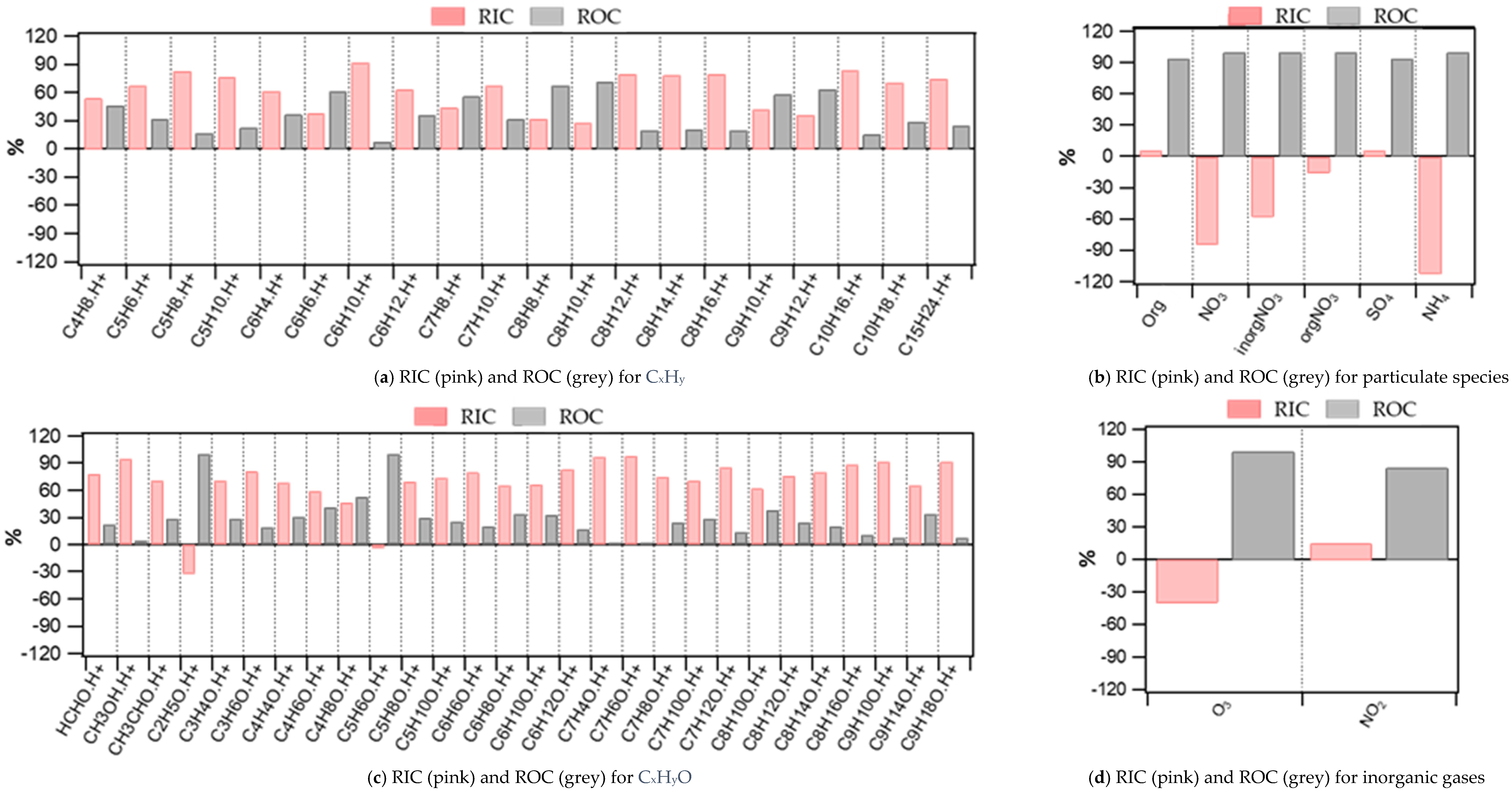
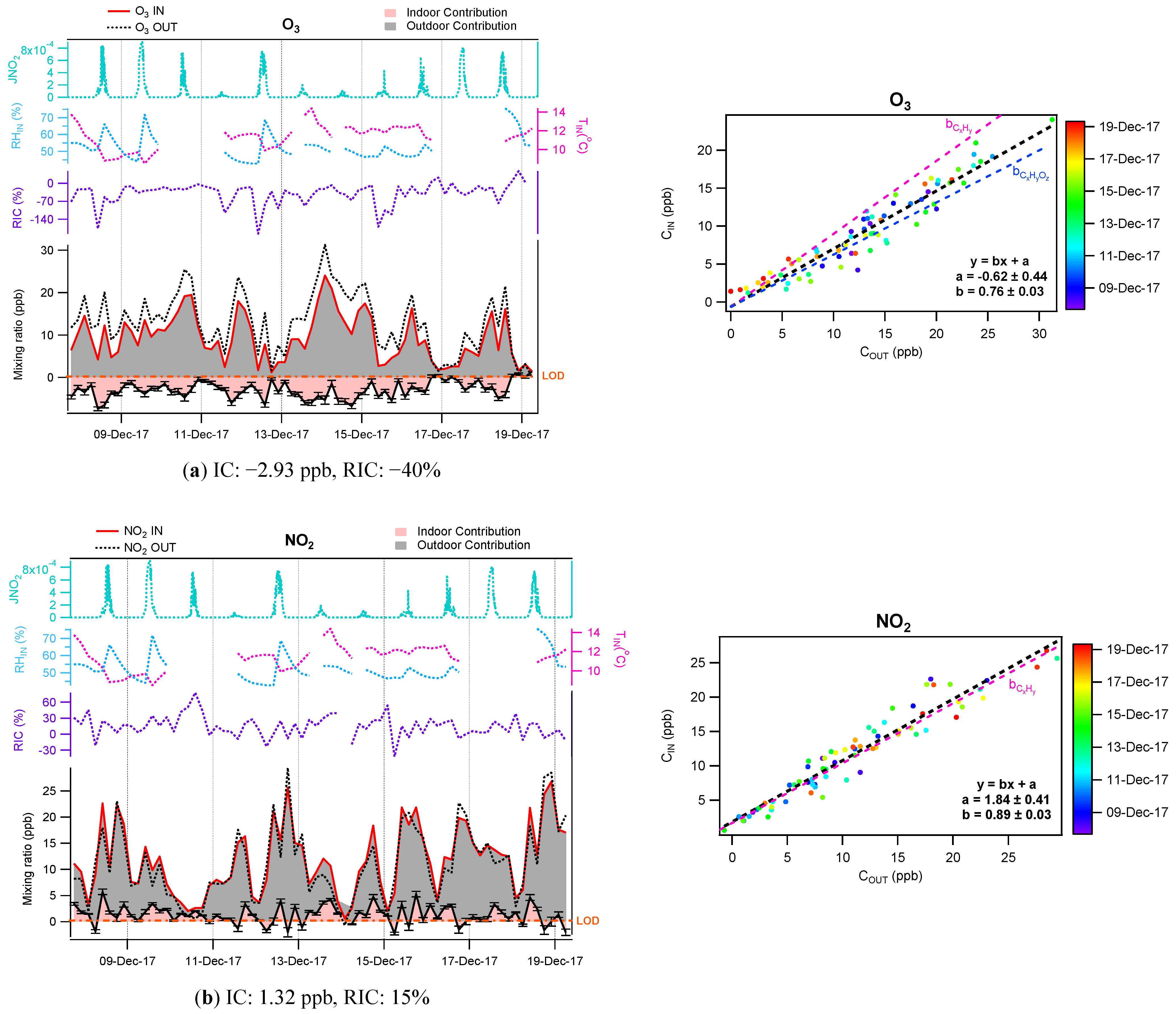
3.2. Contribution of the Indoor Environment to Observed Gaseous Pollutant Concentrations
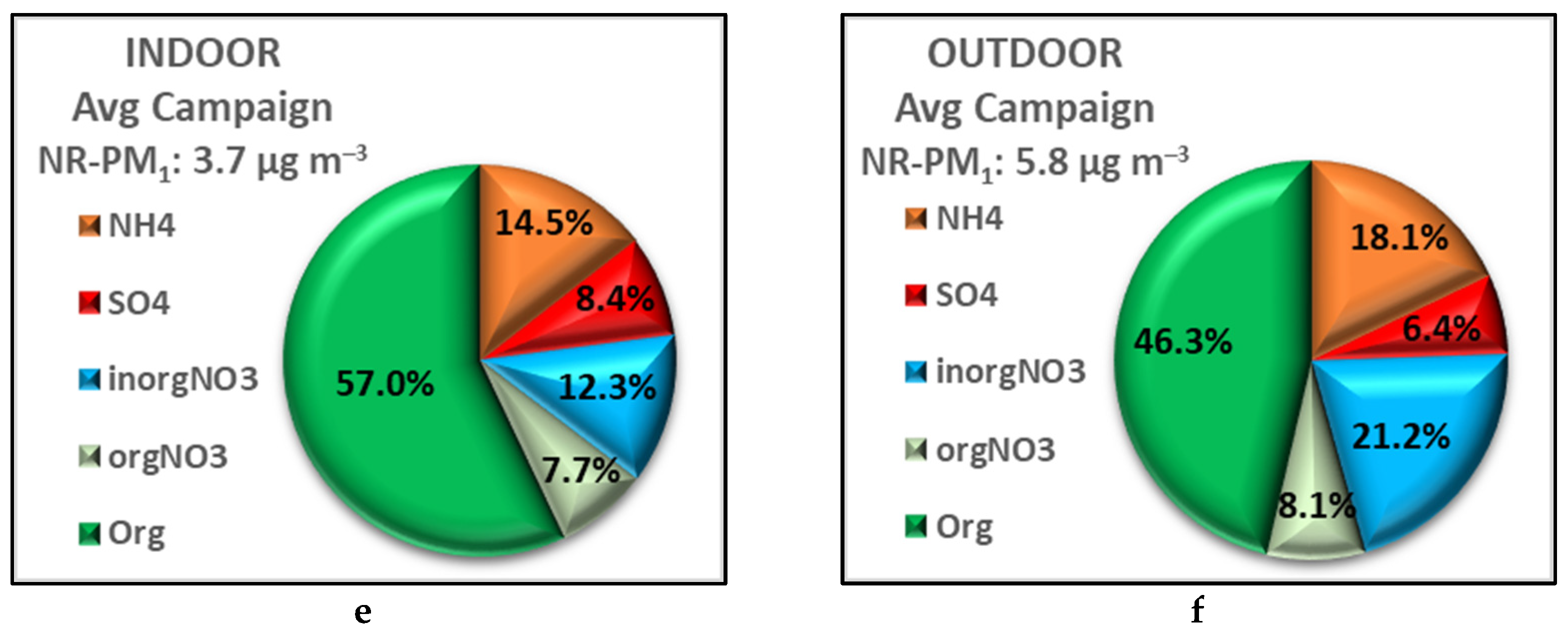
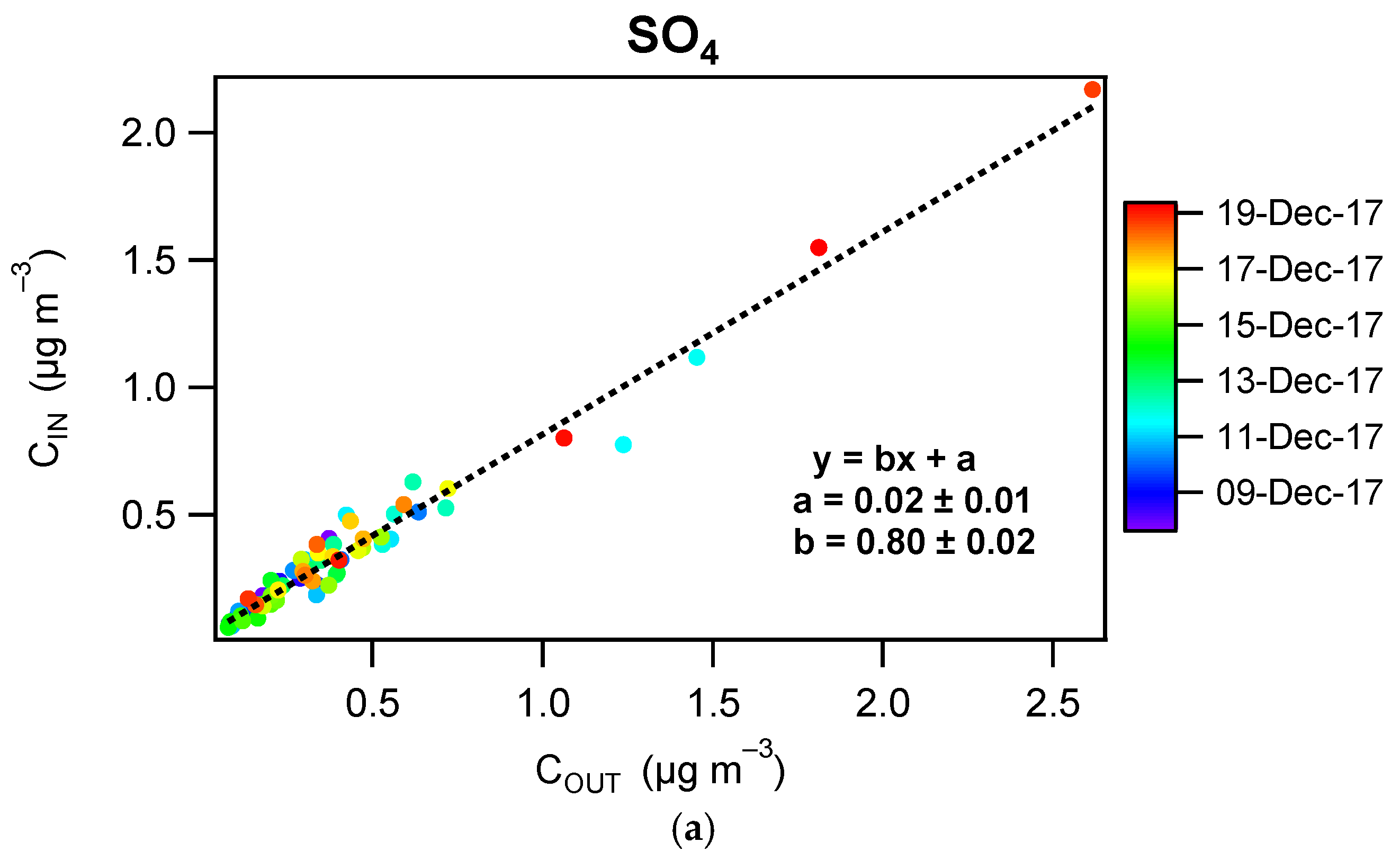
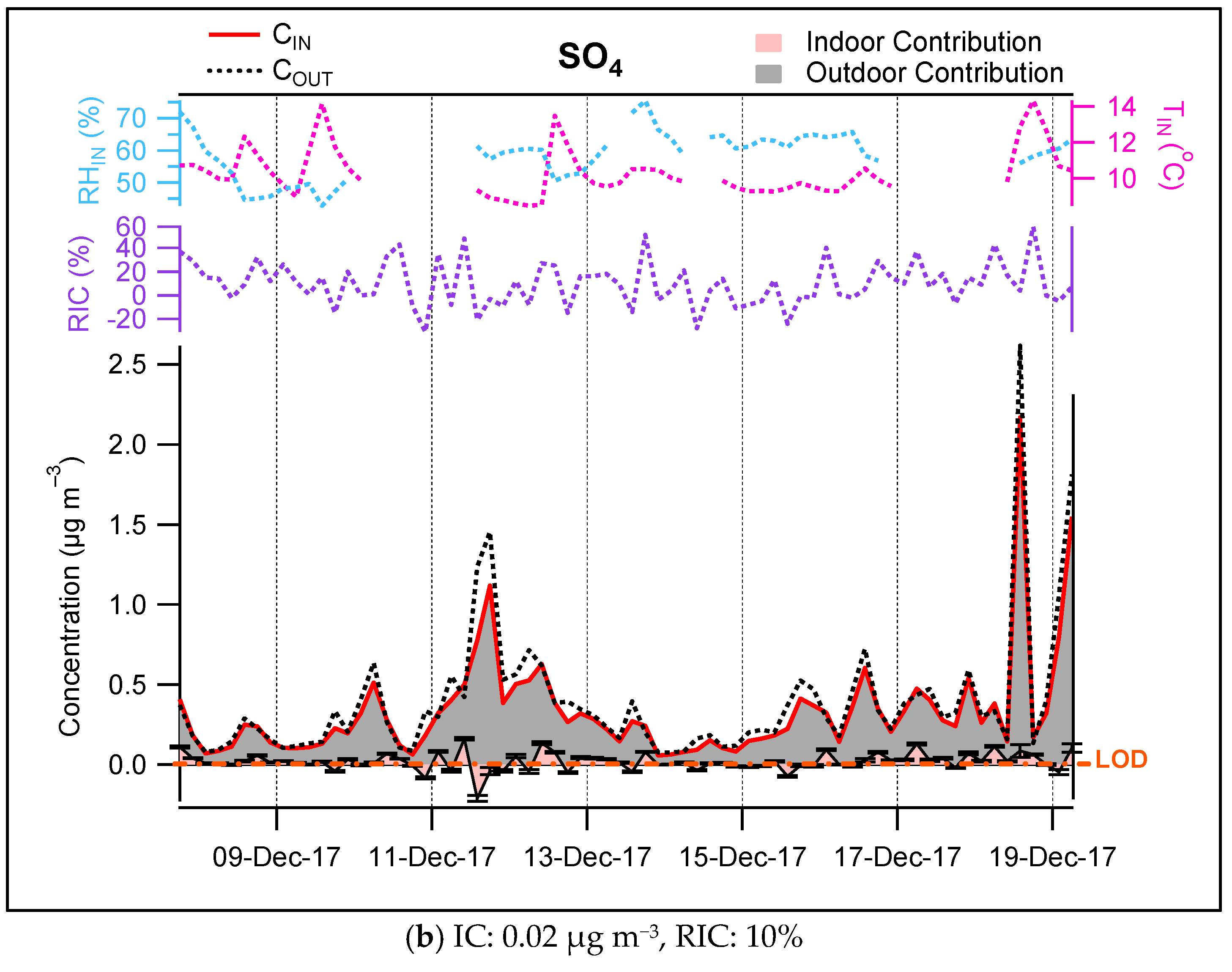
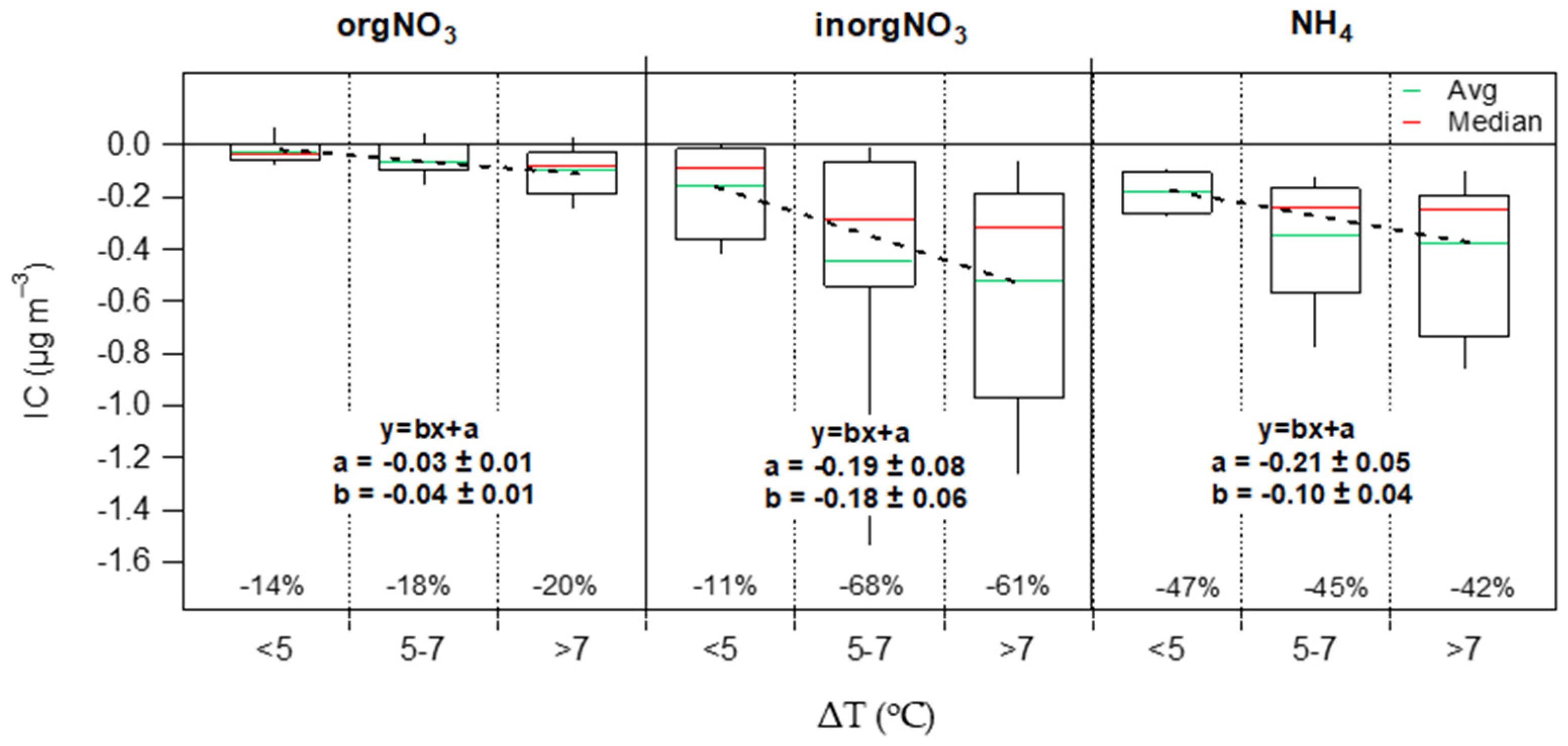
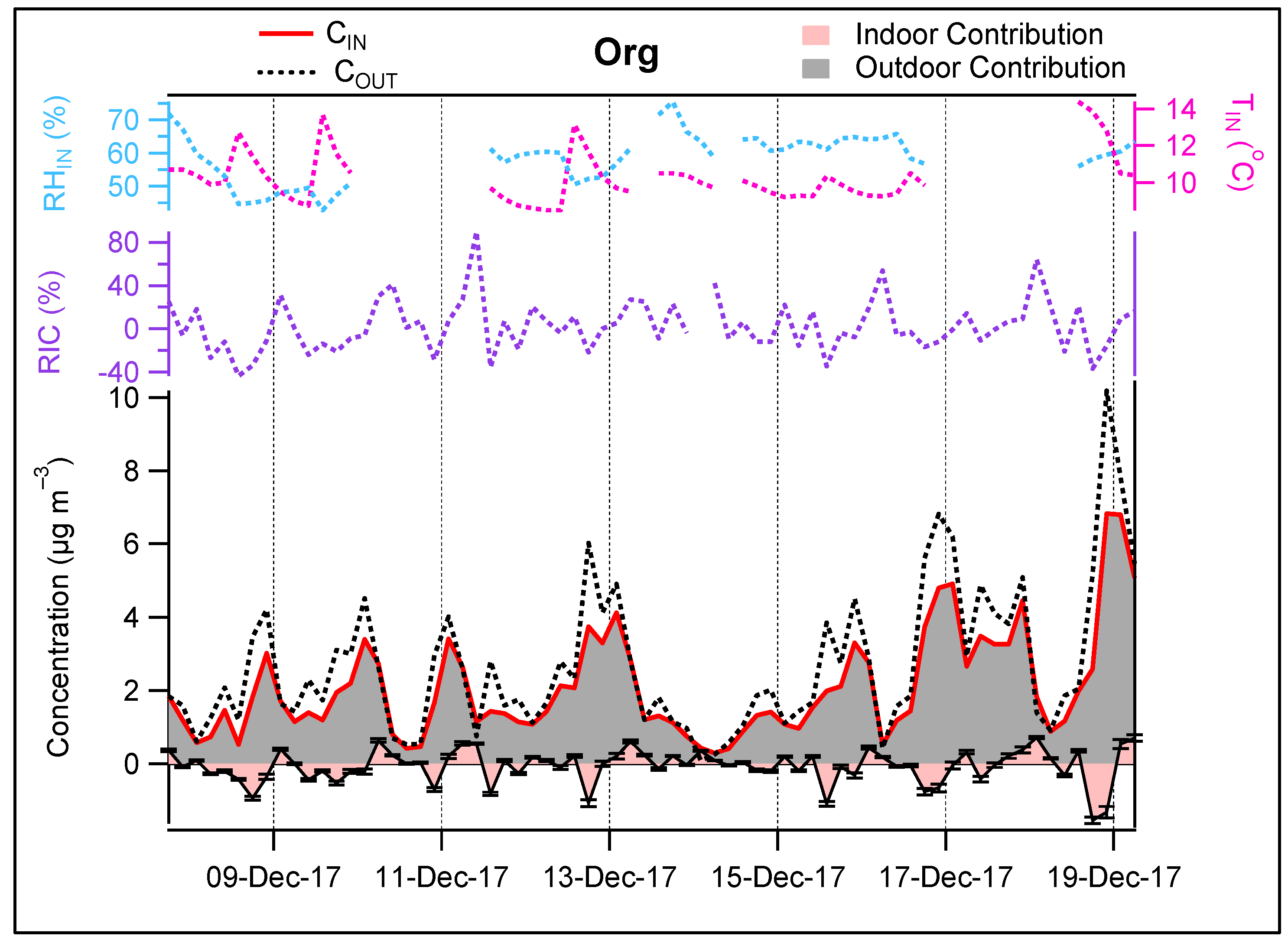
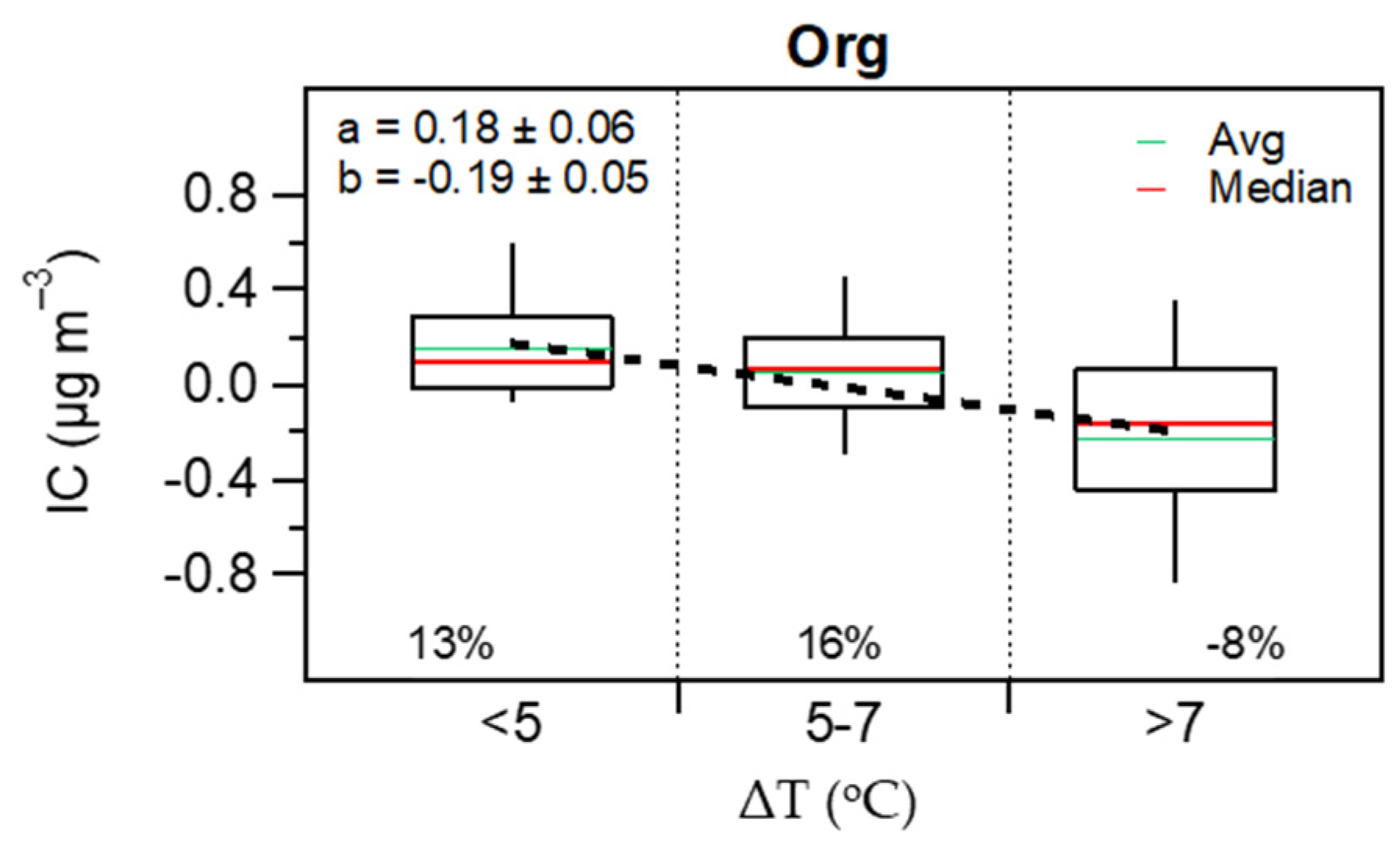
3.3. Contribution of the Indoor Environment to the Observed Concentrations of Submicron Particulate Species
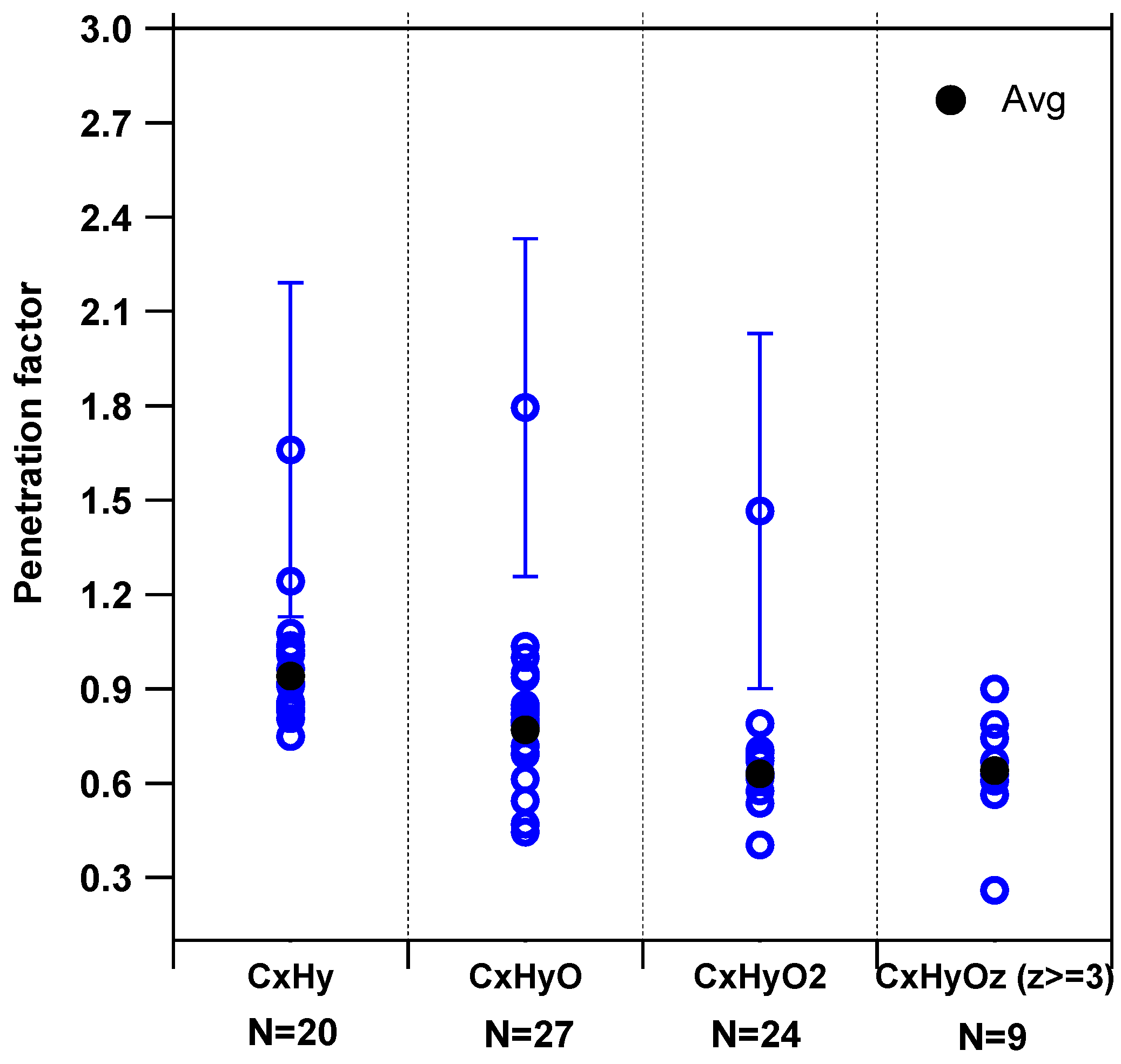
3.4. Method Limitations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wallace, A. The Total Exposure Assessment Methodology (TEAM) Study: An Analysis of Exposures, Sources, and Risks Associated with Four Volatile Organic Chemicals. J. Am. Coll. Toxicol. 1989, 8, 883–895. [Google Scholar] [CrossRef]
- Abbatt, J.P.D.; Wang, C. The atmospheric chemistry of indoor environments. Environ. Sci. Process. Impacts 2020, 22, 25–48. [Google Scholar] [CrossRef] [Green Version]
- Klepeis, N.E.; Nelson, W.C.; Ott, W.R.; Robinson, J.P.; Tsang, A.M.; Switzer, P.; Behar, J.V.; Hern, S.C.; Engelmann, W.H. The National Human Activity Pattern Survey (NHAPS): A resource for assessing exposure to environmental pollutants. J. Expo. Anal. Environ. Epidemiol. 2001, 11, 231–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Burden of Disease from Household Air Pollution for 2016. WHO Regional Office for Europe. 2018. Available online: www.who.int (accessed on 1 December 2021).
- Loomis, D.; Grosse, Y.; Lauby-Secretan, B.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Baan, R.; Mattock, H.; Straif, K. The carcinogenicity of outdoor air pollution. Lancet Oncol. 2013, 14, 1262–1263. [Google Scholar] [CrossRef]
- Kelly, F.J.; Fussell, J.C. Size, source and chemical composition as determinants of toxicity attributable to ambient particulate matter. Atmos. Environ. 2012, 60, 504–526. [Google Scholar] [CrossRef]
- Saleh, Y.; Antherieu, S.; Dusautoir, R.; Alleman, L.Y.; Sotty, J.; De Sousa, C.; Platel, A.; Perdrix, E.; Riffault, V.; Fronval, I.; et al. Exposure to Atmospheric Ultrafine Particles Induces Severe Lung Inflammatory Response and Tissue Remodeling in Mice. Int. J. Environ. Res. Public Health 2019, 16, 1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclercq, B.; Platel, A.; Antherieu, S.; Alleman, L.; Hardy, E.; Perdrix, E.; Grova, N.; Riffault, V.; Appenzeller, B.; Happillon, M.; et al. Genetic and epigenetic alterations in normal and sensitive COPD-diseased human bronchial epithelial cells repeatedly exposed to air pollution-derived PM 2.5. Environ. Pollut. 2017, 230, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Lanzinger, S.; Schneider, A.; Breitner, S.; Stafoggia, M.; Erzen, I.; Dostal, M.; Pastorkova, A.; Bastian, S.; Cyrys, J.; Zscheppang, A.; et al. Associations between ultrafine and fine particles and mortality in five central European cities—Results from the UFIREG study. Environ. Int. 2016, 88, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Oberdörster, G.; Oberdörster, E.; Oberdörster, J. Nanotoxicology: An Emerging Discipline Evolving from Studies of Ultrafine Particles. Environ. Health Perspect. 2005, 113, 823–839. [Google Scholar] [CrossRef]
- Pandis, S.N.; Skyllakou, K.; Florou, K.; Kostenidou, E.; Kaltsonoudis, C.; Hasa, E.; Presto, A.A. Urban particulate matter pollution: A tale of five cities. Faraday Discuss. 2016, 189, 277–290. [Google Scholar] [CrossRef]
- Hu, K.; Guo, Y.; Hu, D.; Du, R.; Yang, X.; Zhong, J.; Fei, F.; Chen, F.; Chen, G.; Zhao, Q.; et al. Mortality burden attributable to PM1 in Zhejiang province, China. Environ. Int. 2018, 121, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Li, S.; Zhang, Y.; Zhang, W.; Li, D.; Wei, X.; He, Y.; Bell, M.L.; Williams, G.; Marks, G.B.; et al. Effects of ambient PM 1 air pollution on daily emergency hospital visits in China: An epidemiological study. Lancet Planet. Health 2017, 1, e221–e229. [Google Scholar] [CrossRef]
- Lin, H.; Tao, J.; Du, Y.; Liu, T.; Qian, Z.; Tian, L.; Di, Q.; Rutherford, S.; Guo, L.; Zeng, W.; et al. Particle size and chemical constituents of ambient particulate pollution associated with cardiovascular mortality in Guangzhou, China. Environ. Pollut. 2016, 208, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Yu, O.; Sheppard, L.; Lumley, T.; Koenig, J.Q.; Shapiro, G.G. Effects of ambient air pollution on symptoms of asthma in Seattle-area children enrolled in the CAMP study. Environ. Health Perspect. 2000, 108, 1209–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwozdziak, A.; Sówka, I.; Willak-Janc, E.; Zwozdziak, J.; Kwiecińska, K.; Balińska-Miśkiewicz, W. Influence of PM1 and PM2.5 on lung function parameters in healthy schoolchildren—A panel study. Environ. Sci. Pollut. Res. 2016, 23, 23892–23901. [Google Scholar] [CrossRef] [PubMed]
- Mar, T.F.; Larson, T.V.; Stier, R.A.; Claiborn, C.; Koenig, J.Q. An Analysis of the Association Between Respiratory Symptoms in Subjects with Asthma and Daily Air Pollution in Spokane, Washington. Inhal. Toxicol. 2004, 16, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Madureira, J.; Paciência, I.; Rufo, J.C.; Severo, M.; Ramos, E.; Barros, H.; Fernandes, E.D.O. Source apportionment of CO2, PM10 and VOCs levels and health risk assessment in naturally ventilated primary schools in Porto, Portugal. Build. Environ. 2016, 96, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, J.A.; Alexis, N.; Bacchus, H.; Bernstein, I.L.; Fritz, P.; Horner, E.; Li, N.; Mason, S.; Nel, A.; Oullette, J.; et al. The health effects of nonindustrial indoor air pollution. J. Allergy Clin. Immunol. 2008, 121, 585–591. [Google Scholar] [CrossRef]
- Wolkoff, P.; Wilkins, C.K.; Clausen, P.A.; Nielsen, G.D. Organic compounds in office environments-sensory irritation, odor, measurements and the role of reactive chemistry. Indoor Air 2005, 16, 7–19. [Google Scholar] [CrossRef]
- Zhang, J.F.; Smith, K.R. Indoor air pollution: A global health concern. Br. Med. Bull. 2003, 68, 209–225. [Google Scholar] [CrossRef] [Green Version]
- Alford, K.L.; Kumar, N. Pulmonary Health Effects of Indoor Volatile Organic Compounds—A Meta-Analysis. Int. J. Environ. Res. Public Health 2021, 18, 1578. [Google Scholar] [CrossRef] [PubMed]
- European Enviromental Agency. Air Quality in Europe. NO 28/2016. Available online: https://www.eea.europa.eu/publications/air-quality-in-europe-2016 (accessed on 1 December 2021).
- Service Public de la Diffusion du Droit. Décret n 2015-1000 du 17 Août 2015 Relatif aux Modalités de Surveillance de la Qualité de l’air Intérieur dans Certains Établissements Recevant du Public; 2015; Available online: https://www.legifrance.gouv.fr/loda/id/JORFTEXT000031052712/2015-08-20#JORFTEXT000031052712 (accessed on 25 March 2022).
- Agence Nationale de Sécurité Sanitaire de L’alimentation. Valeurs Guides de Qualité d’Air Intérieur (VGAI). Available online: https://www.anses.fr/fr/content/valeurs-guides-de-qualit%C3%A9-d%E2%80%99air-int%C3%A9rieur-vgai (accessed on 25 March 2022).
- Avery, A.M.; Waring, M.S.; Decarlo, P.F. Seasonal variation in aerosol composition and concentration upon transport from the outdoor to indoor environment. Environ. Sci. Process. Impacts 2019, 21, 528–547. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.M.; Waring, M.S.; DeCarlo, P.F. Real-time transformation of outdoor aerosol components upon transport indoors measured with aerosol mass spectrometry. Indoor Air 2017, 27, 230–240. [Google Scholar] [CrossRef]
- Bari, M.A.; Kindzierski, W.B.; Wheeler, A.J.; Héroux, M.È.; Wallace, L.A. Source apportionment of indoor and outdoor volatile organic compounds at homes in Edmonton, Canada. Build. Environ. 2015, 90, 114–124. [Google Scholar] [CrossRef]
- Bari, A.; Kindzierski, W.B.; Wallace, L.A.; Wheeler, A.J.; MacNeill, M.; Héroux, M.-È. Indoor and Outdoor Levels and Sources of Submicron Particles (PM1) at Homes in Edmonton, Canada. Environ. Sci. Technol. 2015, 49, 6419–6429. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, R.; Wild, R.J.; Warneke, C.; de Gouw, J.; Brown, S.S.; Miller, S.L.; Luongo, J.C.; Jimenez, J.L.; Ziemann, P.J. Contribution of human-related sources to indoor volatile organic compounds in a university classroom. Indoor Air 2016, 26, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Omelekhina, Y.; Eriksson, A.; Canonaco, F.; Prevot, A.S.H.; Nilsson, P.; Isaxon, C.; Pagels, J.; Wierzbicka, A. Cooking and electronic cigarettes leading to large differences between indoor and outdoor particle composition and concentration measured by aerosol mass spectrometry. Environ. Sci. Process. Impacts 2020, 22, 1382–1396. [Google Scholar] [CrossRef]
- Plaisance, H.; Mocho, P.; Sauvat, N.; Vignau-Laulhere, J.; Raulin, K.; Desauziers, V. Using the chemical mass balance model to estimate VOC source contributions in newly built timber frame houses: A case study. Environ. Sci. Pollut. Res. 2017, 24, 24156–24166. [Google Scholar] [CrossRef]
- Kopperud, R.J.; Ferro, A.R.; Hildemann, L.M. Outdoor Versus Indoor Contributions to Indoor Particulate Matter (PM) Determined by Mass Balance Methods. J. Air Waste Manag. Assoc. 2004, 54, 1188–1196. [Google Scholar] [CrossRef]
- Carslaw, N. A new detailed chemical model for indoor air pollution. Atmos. Environ. 2007, 41, 1164–1179. [Google Scholar] [CrossRef]
- Shiraiwa, M.; Carslaw, N.; Tobias, D.J.; Waring, M.S.; Rim, D.; Morrison, G.; Lakey, P.S.J.; Kruza, M.; Von Domaros, M.; Cummings, B.E.; et al. Modelling consortium for chemistry of indoor environments (MOCCIE): Integrating chemical processes from molecular to room scales. Environ. Sci. Process. Impacts 2019, 21, 1240–1254. [Google Scholar] [CrossRef] [PubMed]
- Chavent, M.; Guégan, H.; Kuentz, V.; Patouille, B.; Saracco, J. PCA- and PMF-based methodology for air pollution sources identification and apportionment. Environmetrics 2009, 20, 928–942. [Google Scholar] [CrossRef] [Green Version]
- RT2012: Un saut énergétique pour les bâtiments neufs. Available online: https://www.ecologie.gouv.fr/reglementation-thermique-rt2012#:~:text=La%20RT2012%20permet%20de%20prendre,confondre%20avec%20l’%C3%A9nergie%20finale (accessed on 25 March 2022).
- Guerrero, F. ASTM E 741—00: Standard Test Method for Determining Air Change in a Single Zone by Means of a Tracer Gas Dilution. 200. Available online: https://www.academia.edu/40099720/Standard_Test_Method_for_Determining_Air_Change_in_a_Single_Zone_by_Means_of_a_Tracer_Gas_Dilution_1 (accessed on 25 March 2022).
- Stratigou, E.; Dusanter, S.; Brito, J.; Riffault, V. Investigation of PM10, PM2.5, PM1 in an unoccupied airflow-controlled room: How reliable to neglect resuspension and assume unreactive particles? Build. Environ. 2020, 186, 107357. [Google Scholar] [CrossRef]
- DeCarlo, P.F.; Kimmel, J.R.; Trimborn, A.; Northway, M.J.; Jayne, J.T.; Aiken, A.C.; Gonin, M.; Fuhrer, K.; Horvath, T.; Docherty, K.S.; et al. Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer. Anal. Chem. 2006, 78, 8281–8289. [Google Scholar] [CrossRef]
- Canagaratna, M.; Jayne, J.; Jimenez, J.; Allan, J.; Alfarra, M.; Zhang, Q.; Onasch, T.; Drewnick, F.; Coe, H.; Middlebrook, A.; et al. Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer. Mass Spectrom. Rev. 2007, 26, 185–222. [Google Scholar] [CrossRef]
- Crenn, V.; Fronval, I.; Petitprez, D.; Riffault, V. Fine particles sampled at an urban background site and an industrialized coastal site in Northern France—Part 1: Seasonal variations and chemical characterization. Sci. Total Environ. 2017, 578, 203–218. [Google Scholar] [CrossRef]
- Lindinger, W.; Hansel, A.; Jordan, A. On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) medical applications, food control and environmental research. Int. J. Mass Spectrom. Ion Process. 1998, 173, 191–241. [Google Scholar] [CrossRef]
- de Gouw, J.; Warneke, C.; Karl, T.; Eerdekens, G.; van der Veen, C.; Fall, R. Sensitivity and specificity of atmospheric trace gas detection by proton-transfer-reaction mass spectrometry. Int. J. Mass Spectrom. 2003, 223–224, 365–382. [Google Scholar] [CrossRef]
- Farmer, D.K.; Matsunaga, A.; Docherty, K.S.; Surratt, J.D.; Seinfeld, J.H.; Ziemann, P.J.; Jimenez, J.L. Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry. Proc. Natl. Acad. Sci. USA 2010, 107, 6670–6675. [Google Scholar] [CrossRef] [Green Version]
- Kiendler-Scharr, A.; Mensah, A.; Friese, E.; Topping, D.; Nemitz, E.; Prevot, A.S.H.; Äijälä, M.; Allan, J.; Canonaco, F.; Canagaratna, M.; et al. Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol. Geophys. Res. Lett. 2016, 43, 7735–7744. [Google Scholar] [CrossRef] [Green Version]
- Polidori, A.; Turpin, B.; Meng, Q.Y.; Lee, J.H.; Weisel, C.; Morandi, M.; Colome, S.; Stock, T.; Winer, A.; Zhang, J.; et al. Fine organic particulate matter dominates indoor-generated PM2.5 in RIOPA homes. J. Expo. Sci. Environ. Epidemiol. 2006, 16, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arata, C.; Zarzana, K.J.; Misztal, P.K.; Liu, Y.; Brown, S.S.; Nazaroff, W.W.; Goldstein, A.H. Measurement of NO3 and N2O5 in a Residential Kitchen. Environ. Sci. Technol. Lett. 2018, 5, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Carslaw, N.; Shaw, D. Secondary product creation potential (SPCP): A metric for assessing the potential impact of indoor air pollution on human health. Environ. Sci. Process. Impacts 2019, 21, 1313–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waring, M.S.; Wells, J.R. Volatile organic compound conversion by ozone, hydroxyl radicals, and nitrate radicals in residential indoor air: Magnitudes and impacts of oxidant sources. Atmos. Environ. 2015, 106, 382–391. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, E.G.; Amedro, D.; Afif, C.; Gligorovski, S.; Schoemaecker, C.; Fittschen, C.; Doussin, J.-F.; Wortham, H. Unexpectedly high indoor hydroxyl radical concentrations associated with nitrous acid. Proc. Natl. Acad. Sci. USA 2013, 110, 13294–13299. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, A.T.; Rudd, A.F.; Beal, D.; Chandra, S. Volatile Organic Compound Concentrations and Emission Rates in New Manufactured and Site-Built Houses. Indoor Air 2001, 10, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Holøs, S.B.; Yang, A.; Lind, M.; Thunshelle, K.; Schild, P.; Mysen, M. VOC emission rates in newly built and renovated buildings, and the influence of ventilation—A review and meta-analysis. Int. J. Vent. 2018, 18, 153–166. [Google Scholar] [CrossRef]
- Villanueva, F.; Tapia, A.; Lara, S.; Amo-Salas, M. Indoor and outdoor air concentrations of volatile organic compounds and NO2 in schools of urban, industrial and rural areas in Central-Southern Spain. Sci. Total Environ. 2018, 622–623, 222–235. [Google Scholar] [CrossRef]
- Fortenberry, C.; Walker, M.; Dang, A.; Loka, A.; Date, G.; De Carvalho, K.C.; Morrison, G.; Williams, B. Analysis of indoor particles and gases and their evolution with natural ventilation. Indoor Air 2019, 29, 761–779. [Google Scholar] [CrossRef]
- Liu, S.; Thompson, S.L.; Stark, H.; Ziemann, P.J.; Jimenez, J.L. Gas-Phase Carboxylic Acids in a University Classroom: Abundance, Variability, and Sources. Environ. Sci. Technol. 2017, 51, 5454–5463. [Google Scholar] [CrossRef]
- Patkó, C.; Patko, I.; Pásztory, Z. Indoor Air Quality Testing in Low-Energy Wooden Houses: Measurement of Formaldehyde and VOC-s. Acta Polytech. Hung. 2013, 10, 105–116. [Google Scholar]
- Derbez, M.; Berthineau, B.; Cochet, V.; Lethrosne, M.; Pignon, C.; Riberon, J.; Kirchner, S. Indoor air quality and comfort in seven newly built, energy-efficient houses in France. Build. Environ. 2014, 72, 173–187. [Google Scholar] [CrossRef]
- Solomon, S.; Schade, G.; Kuttippurath, J.; Ladstätter-Weissenmayer, A.; Burrows, J.P. VOC Concentrations in an Indoor Workplace Environment of a University Building. Indoor Built Environ. 2008, 17, 260–268. [Google Scholar] [CrossRef]
- Allen, J.G.; Macnaughton, P.; Satish, U.; Santanam, S.; Vallarino, J.; Spengler, J.D. Associations of Cognitive Function Scores with Carbon Dioxide, Ventilation, and Volatile Organic Compound Exposures in Office Workers: A Controlled Exposure Study of Green and Conventional Office Environments. Environ. Health Perspect. 2016, 124, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.-H.; Jo, W.-K. Longitudinal variations in indoor VOC concentrations after moving into new apartments and indoor source characterization. Environ. Sci. Pollut. Res. 2013, 20, 3696–3707. [Google Scholar] [CrossRef]
- Schoemaecker, C.; Verriele, M.; Hanoun, B.; Petitprez, D.; Leclerc, N.; Dusanter, S.; Le Calve, S.; Millet, M.; Bernhardt, P.; Mendez, M.; et al. Characterization of the IAQ in Low Energy Public Buildings in France through a Dual Experiment and Modeling Approach; ICPEES: Hiltigham, France, 2014. [Google Scholar]
- Geiss, O.; Giannopoulos, G.; Tirendi, S.; Barrero-Moreno, J.; Larsen, B.R.; Kotzias, D. The AIRMEX study-VOC measurements in public buildings and schools/kindergartens in eleven European cities: Statistical analysis of the data. Atmos. Environ. 2011, 45, 3676–3684. [Google Scholar] [CrossRef]
- Csobod, E.; Annesi-Maesano, I.; Carrer, P.; Kephalopoulos, S.; Madureira, J.; Rudnai, P.; De Oliveira, F.E.; Barrero, J.; Beregszászi, T.; Hyvärinen, A.; et al. SINPHONIE: Schools Indoor Pollution & Health Observatory Network in Europe; EUR—Scientific and Technical Research Reports; Publications Office of the European Union: Luxembourg, 2014. [Google Scholar]
- Schlink, U.; Rehwagen, M.; Damm, M.; Richter, M.; Borte, M.; Herbarth, O. Seasonal cycle of indoor-VOCs: Comparison of apartments and cities. Atmos. Environ. 2004, 38, 1181–1190. [Google Scholar] [CrossRef]
- Tang, X.; Misztal, P.K.; Nazaroff, W.W.; Goldstein, A.H. Volatile Organic Compound Emissions from Humans Indoors. Environ. Sci. Technol. 2016, 50, 12686–12694. [Google Scholar] [CrossRef] [Green Version]
- Duncan, S.M.; Tomaz, S.; Morrison, G.; Webb, M.; Atkin, J.M.; Surratt, J.D.; Turpin, B.J. Dynamics of Residential Water-Soluble Organic Gases: Insights into Sources and Sinks. Environ. Sci. Technol. 2019, 53, 1812–1821. [Google Scholar] [CrossRef]
- Dodson, R.E.; Levy, J.I.; Spengler, J.D.; Shine, J.P.; Bennett, D.H. Influence of basements, garages, and common hallways on indoor residential volatile organic compound concentrations. Atmos. Environ. 2008, 42, 1569–1581. [Google Scholar] [CrossRef]
- Jia, C.; Batterman, S.; Godwin, C.; Charles, S.M.; Chin, J.-Y. Sources and migration of volatile organic compounds in mixed-use buildings. Indoor Air 2010, 20, 357–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smedemark, S.H.; Ryhl-Svendsen, M.; Schieweck, A. Quantification of formic acid and acetic acid emissions from heritage collections under indoor room conditions. Part I: Laboratory and field measurements. Heritage Sci. 2020, 8, 58. [Google Scholar] [CrossRef]
- Zhang, J.; Wilson, E.W.; Lioy, P.J. Sources of organic acids in indoor air: A field study. J. Expo. Anal. Environ. Epidemiol. 1994, 4, 25–47. [Google Scholar] [PubMed]
- Talbot, N.; Kubelová, L.; Makeš, O.; Ondráček, J.; Cusack, M.; Schwarz, J.; Vodička, P.; Zíková, N.; Ždímal, V. Transformations of Aerosol Particles from an Outdoor to Indoor Environment. Aerosol Air Qual. Res. 2017, 17, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Duncan, S.M.; Sexton, K.G.; Turpin, B.J. Oxygenated VOCs, aqueous chemistry, and potential impacts on residential indoor air composition. Indoor Air 2017, 28, 198–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Misztal, P.K.; Xiong, J.; Tian, Y.; Arata, C.; Weber, R.J.; Nazaroff, W.W.; Goldstein, A.H. Characterizing sources and emissions of volatile organic compounds in a northern California residence using space- and time-resolved measurements. Indoor Air 2019, 29, 630–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.; Langer, S.; Ljungström, E. Chemistry and indoor air quality in a multi-storey wooden passive (low energy) building: Formation of peroxyacetyl nitrate. Indoor Built Environ. 2013, 23, 485–496. [Google Scholar] [CrossRef]
- Offermann, F.J.; Maddalena, R.L.; Offermann, F.J.; Singer, B.C.; Willem, H. The impact of ventilation rate on the emission rates of volatile organic compounds in residences. In Proceedings of the Healthy Buildings 2012—10th International Conference, Brisbane, Australia, 8–12 July 2012. [Google Scholar]
- Finlayson-Pitts, B.J.; Pitts, J.N., Jr. Chemistry of the Upper and Lower Atmosphere Theory, Experiments, and Applications; Elsevier: Amsterdam, The Netherlands, 2000. [Google Scholar]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Instrument (Manufacturer) | Measured Parameters or Species | Time Resolution | LOD (3σ) | Location | |
|---|---|---|---|---|---|
| AEROTRAK Handheld Particle Counter 8220 (TSI) | Particle number concentrations (6 size bins: 0.3–5.0 μm) | 50 s | n/a | IN, OUT | |
| HR-ToF-AMS (Aerodyne Research) | Non-refractory PM1: NO3, SO4, Cl, NH4 and Organics | 5 min | Species * | V mode (μg m−3) | IN, OUT |
| Org | 0.64 | ||||
| SO4 | 0.05 | ||||
| NO3 | 0.28 | ||||
| NH4 | 0.37 | ||||
| Cl | 0.19 | ||||
| PTR—QiToFMS (Ionicon Analytik) | VOCs | 10 s | m/z 79: <30 ppt m/z 205: <1 ppt | IN, OUT | |
| CO2 analyzer (Horiba) | CO2 | 1 min | 0.2 ppb | IN, OUT | |
| NO2 CAPS (Aerodyne Research) | NO2 | 10 s | 0.1 ppb | IN, OUT | |
| O3 analyzer (Environnement SA) | O3 | 10 s | 0.4 ppb | IN, OUT | |
| CO2, RH, T probes (Testo) | CO2, Relative Humidity, Temperature | 20 s | n/a | IN | |
| TEOM FDMS (Thermo Fisher Scientific, Waltham, MA, USA) | PM1 mass concentrations | 1 h | n/a | OUT | |
| SMPS (TSI, model 3788) | Particle number concentrations (10.2–414.2 nm) | 4.5 min | n/a | IN, OUT | |
| Spectroradiometer (METCON) | Photolysis rate of NO2 (JNO2) | 1 s | n/a | IN | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stratigou, E.; Dusanter, S.; Brito, J.; Tison, E.; Riffault, V. Using Real Time Measurements to Derive the Indoor and Outdoor Contributions of Submicron Particulate Species and Trace Gases. Toxics 2022, 10, 161. https://doi.org/10.3390/toxics10040161
Stratigou E, Dusanter S, Brito J, Tison E, Riffault V. Using Real Time Measurements to Derive the Indoor and Outdoor Contributions of Submicron Particulate Species and Trace Gases. Toxics. 2022; 10(4):161. https://doi.org/10.3390/toxics10040161
Chicago/Turabian StyleStratigou, Evdokia, Sébastien Dusanter, Joel Brito, Emmanuel Tison, and Véronique Riffault. 2022. "Using Real Time Measurements to Derive the Indoor and Outdoor Contributions of Submicron Particulate Species and Trace Gases" Toxics 10, no. 4: 161. https://doi.org/10.3390/toxics10040161
APA StyleStratigou, E., Dusanter, S., Brito, J., Tison, E., & Riffault, V. (2022). Using Real Time Measurements to Derive the Indoor and Outdoor Contributions of Submicron Particulate Species and Trace Gases. Toxics, 10(4), 161. https://doi.org/10.3390/toxics10040161