
Indoor Secondary Pollutants Cannot Be Ignored: Third-Hand Smoke



Abstract
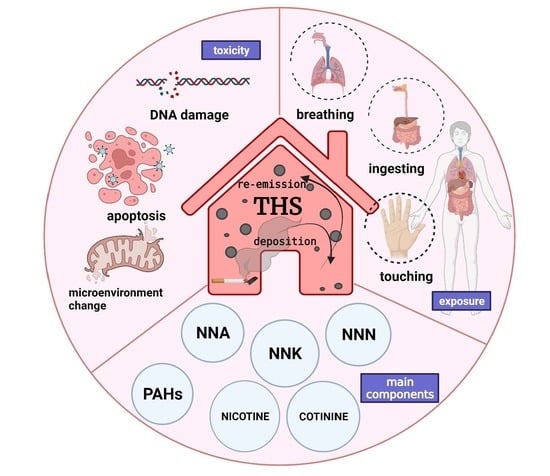
:
1. Introduction
2. Components in THS
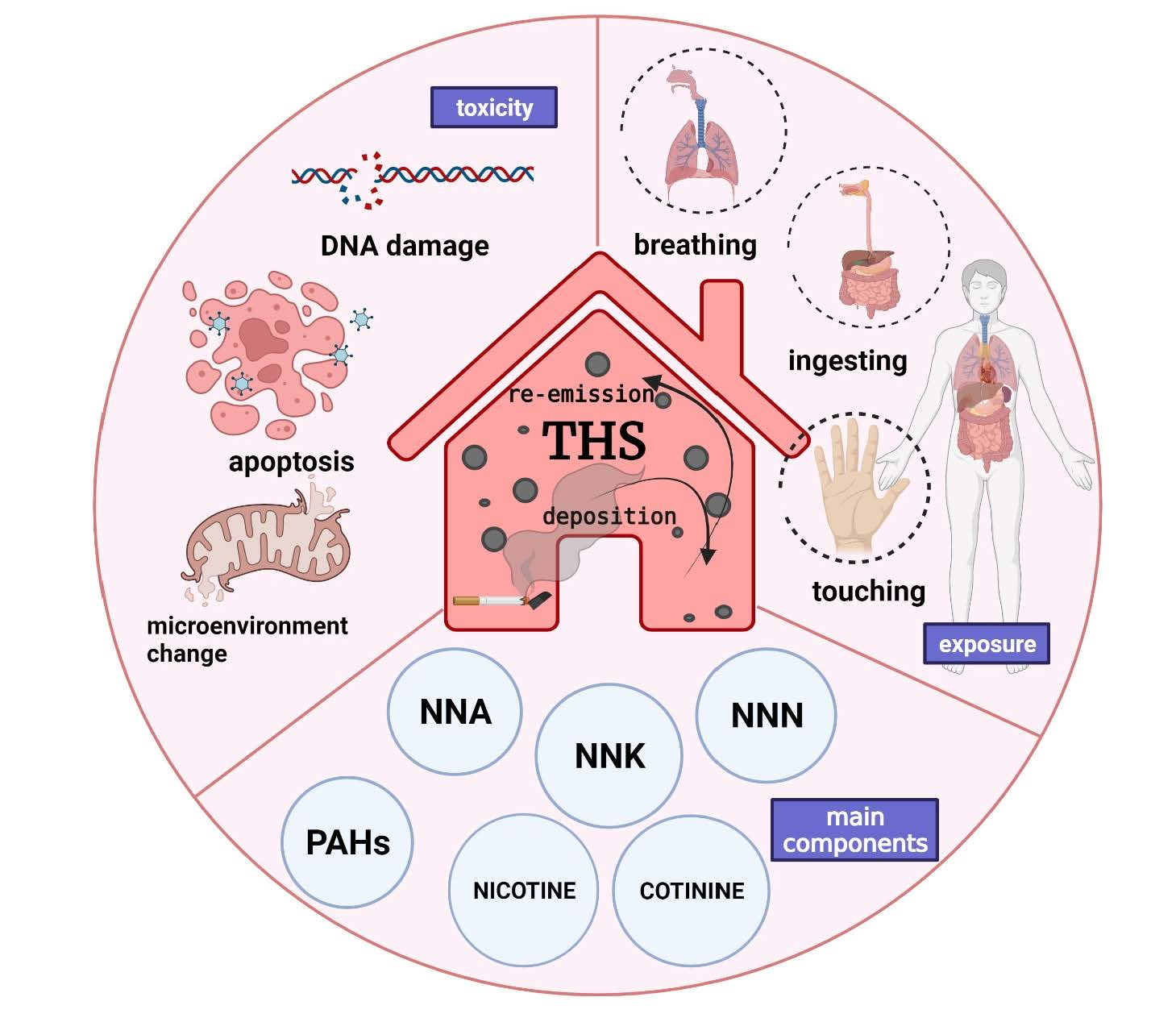
3. Generation of THS
- Selective adsorption and accumulation of certain compounds, such as SVOCs, from the gas phase of SHS onto the surface of objects before they are slowly released into the air;
- PM from SHS stains furniture, walls, carpets, clothing, etc., which can then again be suspended into the air;
- Certain compounds in SHS, such as nicotine, attach to the surface of objects and react with O3, HONO, and other substances in the atmosphere to form second-generation toxicants.
4. Places Where THS Exists
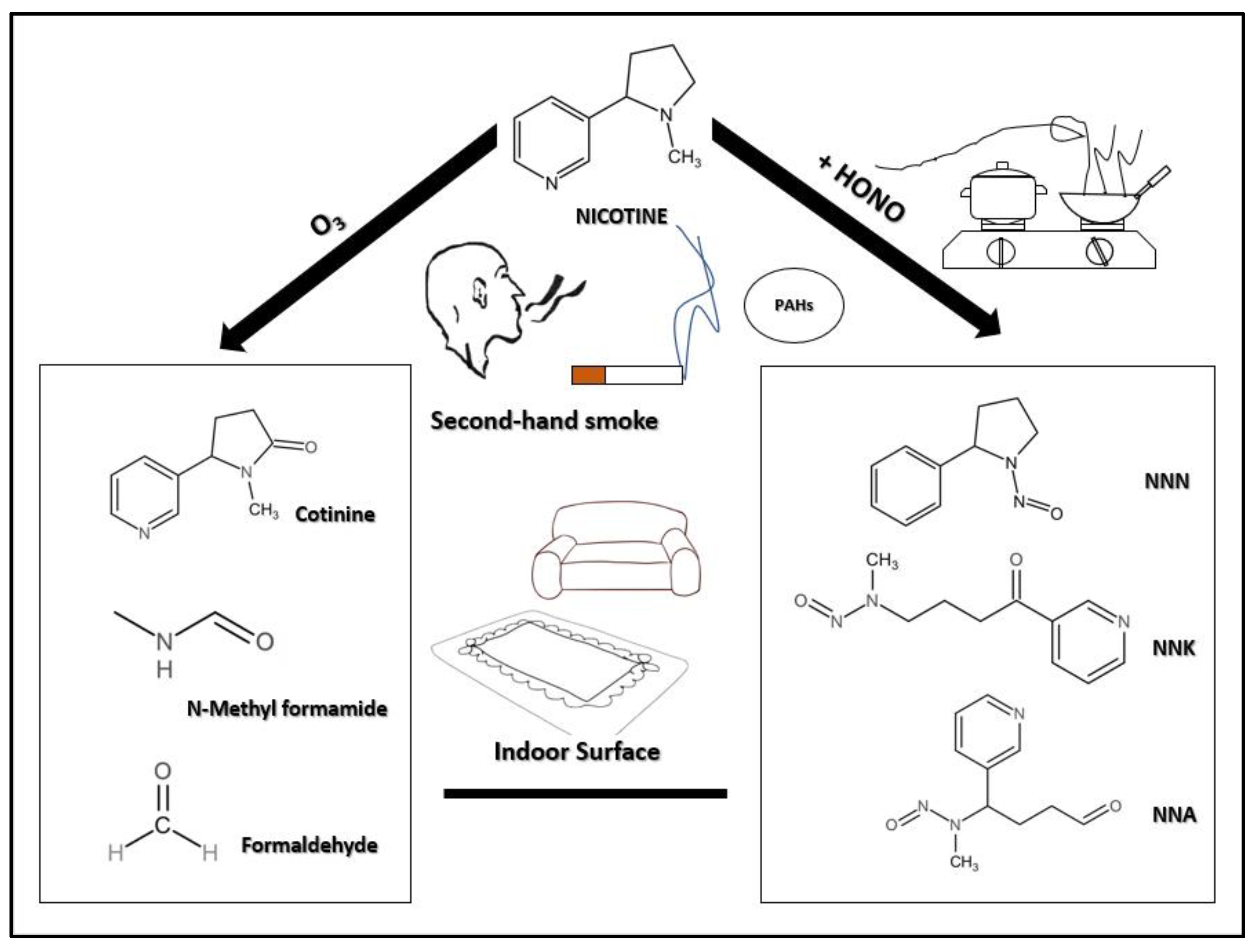
5. The Main Carcinogenic Components of THS
5.1. PAHs: The Killer in Cigarette Smoke
5.2. TSNAs: Products of the Reaction between Nicotine and HONO
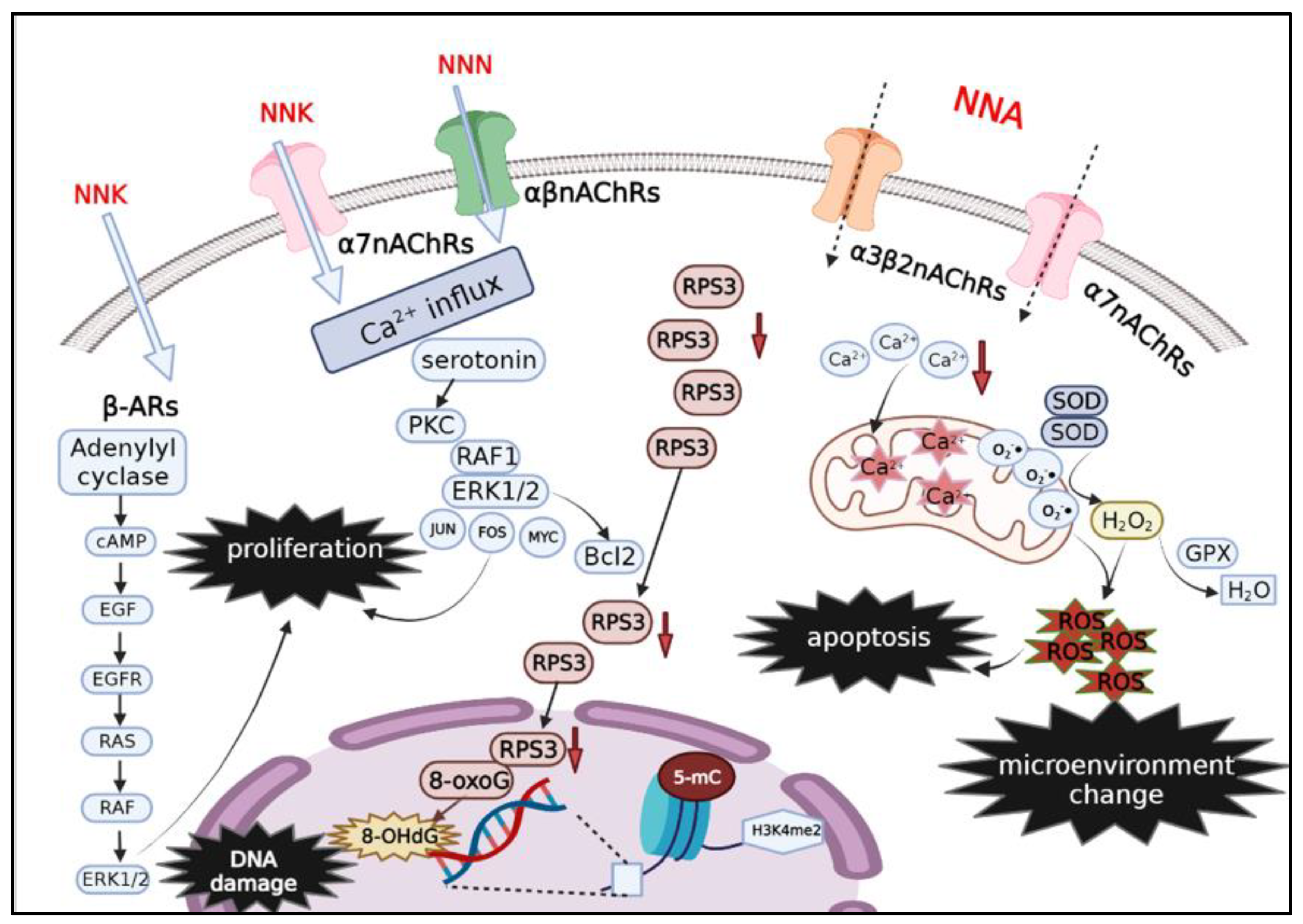
6. Health Effects of THS
7. THS Detection
8. Biomarkers of THS
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Environmental Health Association. Available online: https://www.neha.org/ (accessed on 15 May 2022).
- World Health Organization. Available online: https://www.who.int/ (accessed on 15 May 2022).
- Wang, C.; Collins, D.B. Exploring Conditions for Ultrafine Particle Formation from Oxidation of Cigarette Smoke in Indoor Environments. Environ. Sci. Technol. 2018, 52, 4623–4631. [Google Scholar] [CrossRef] [PubMed]
- Winickoff, J.P.; Friebely, J.; Tanski, S.E.; Sherrod, C.; Matt, G.E.; Hovell, M.F.; McMillen, R.C. Beliefs about the health effects of “thirdhand” smoke and home smoking bans. Pediatrics 2009, 123, e74–e79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centers for Disease Control and Prevention. Tobacco. Available online: http://www.cdc.gov/tobacco (accessed on 15 May 2022).
- China CDC. Available online: https://www.chinacdc.cn/ (accessed on 15 May 2022).
- U.S. Department of Health and Human Services. Available online: https://www.hhs.gov/guidance/document/smoking-and-tobacco-use-cessation-counseling-services-common-working-file-cwf-inquiry (accessed on 28 June 2022).
- Schick, S.F.; Farraro, K.F.; Perrino, C.; Sleiman, M.; van de Vossenberg, G.; Trinh, M.P.; Hammond, S.K.; Jenkins, B.M.; Balmes, J. Thirdhand cigarette smoke in an experimental chamber: Evidence of surface deposition of nicotine, nitrosamines and polycyclic aromatic hydrocarbons and de novo formation of NNK. Tob. Control 2014, 23, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, M.; Gundel, L.A.; Pankow, J.F.; Jacob, P., 3rd; Singer, B.C.; Destaillats, H. Formation of carcinogens indoors by surface-mediated reactions of nicotine with nitrous acid, leading to potential thirdhand smoke hazards. Proc. Natl. Acad. Sci. USA 2010, 107, 6576–6581. [Google Scholar] [CrossRef] [Green Version]
- Sleiman, M.; Logue, J.M.; Luo, W.; Pankow, J.F.; Gundel, L.A.; Destaillats, H. Inhalable constituents of thirdhand tobacco smoke: Chemical characterization and health impact considerations. Environ. Sci. Technol. 2014, 48, 13093–13101. [Google Scholar] [CrossRef] [Green Version]
- Hoh, E.; Hunt, R.N.; Quintana, P.J.; Zakarian, J.M.; Chatfield, D.A.; Wittry, B.C.; Rodriguez, E.; Matt, G.E. Environmental tobacco smoke as a source of polycyclic aromatic hydrocarbons in settled household dust. Environ. Sci. Technol. 2012, 46, 4174–4183. [Google Scholar] [CrossRef]
- Ramírez, N.; Vallecillos, L.; Lewis, A.C.; Borrull, F.; Marcé, R.M.; Hamilton, J.F. Comparative study of comprehensive gas chromatography-nitrogen chemiluminescence detection and gas chromatography-ion trap-tandem mass spectrometry for determining nicotine and carcinogen organic nitrogen compounds in thirdhand tobacco smoke. J. Chromatogr. A 2015, 1426, 191–200. [Google Scholar] [CrossRef]
- Ramírez, N.; Özel, M.Z.; Lewis, A.C.; Marcé, R.M.; Borrull, F.; Hamilton, J.F. Exposure to nitrosamines in thirdhand tobacco smoke increases cancer risk in non-smokers. Environ. Int. 2014, 71, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Daisey, J.M. Tracers for assessing exposure to environmental tobacco smoke: What are they tracing? Environ. Health Perspect. 1999, 107 (Suppl. S2), 319–327. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Nematollahi, M.; Sextro, R.G.; Gadgil, A.J.; Nazaroff, W.W. Deposition of Tobacco Smoke Particles in a Low Ventilation Room. Aerosol. Sci. Technol. 1994, 20, 194–206. [Google Scholar] [CrossRef]
- Destaillats, H.; Singer, B.C.; Lee, S.K.; Gundel, L.A. Effect of ozone on nicotine desorption from model surfaces: Evidence for heterogeneous chemistry. Environ. Sci. Technol. 2006, 40, 1799–1805. [Google Scholar] [CrossRef] [Green Version]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Tobacco smoke and involuntary smoking. IARC Monogr. Eval. Carcinog. Risks Hum. 2004, 83, 1–1438. [Google Scholar]
- Gerde, P.; Muggenburg, B.A.; Thornton-Manning, J.R.; Lewis, J.L.; Pyon, K.H.; Dahl, A.R. Benzo[a]pyrene at an environmentally relevant dose is slowly absorbed by, and extensively metabolized in, tracheal epithelium. Carcinogenesis 1997, 18, 1825–1832. [Google Scholar] [CrossRef] [Green Version]
- Deutsch-Wenzel, R.P.; Brune, H.; Grimmer, G.; Dettbarn, G.; Misfeld, J. Experimental studies in rat lungs on the carcinogenicity and dose-response relationships of eight frequently occurring environmental polycyclic aromatic hydrocarbons. J. Natl. Cancer Inst. 1983, 71, 539–544. [Google Scholar]
- Wolterbeek, A.P.; Schoevers, E.J.; Rutten, A.A.; Feron, V.J. A critical appraisal of intratracheal instillation of benzo[a]pyrene to Syrian golden hamsters as a model in respiratory tract carcinogenesis. Cancer Lett. 1995, 89, 107–116. [Google Scholar] [CrossRef]
- Greenblatt, M.S.; Bennett, W.P.; Hollstein, M.; Harris, C.C. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994, 54, 4855–4878. [Google Scholar]
- Alexandrov, K.; Rojas, M.; Geneste, O.; Castegnaro, M.; Camus, A.M.; Petruzzelli, S.; Giuntini, C.; Bartsch, H. An improved fluorometric assay for dosimetry of benzo(a)pyrene diol-epoxide-DNA adducts in smokers’ lung: Comparisons with total bulky adducts and aryl hydrocarbon hydroxylase activity. Cancer Res. 1992, 52, 6248–6253. [Google Scholar]
- Gao, M.; Li, Y.; Sun, Y.; Long, J.; Kong, Y.; Yang, S.; Wang, Y. A common carcinogen benzo[a]pyrene causes p53 overexpression in mouse cervix via DNA damage. Mutat. Res. 2011, 724, 69–75. [Google Scholar] [CrossRef]
- Gao, M.; Li, Y.; Ji, X.; Xue, X.; Chen, L.; Feng, G.; Zhang, H.; Wang, H.; Shah, W.; Hou, Z.; et al. Disturbance of Bcl-2, Bax, Caspase-3, Ki-67 and C-myc expression in acute and subchronic exposure to benzo(a)pyrene in cervix. Acta Histochem. 2016, 118, 63–73. [Google Scholar] [CrossRef]
- He, J.; Ji, X.; Li, Y.; Xue, X.; Feng, G.; Zhang, H.; Wang, H.; Gao, M. Subchronic exposure of benzo(a)pyrene interferes with the expression of Bcl-2, Ki-67, C-myc and p53, Bax, Caspase-3 in sub-regions of cerebral cortex and hippocampus. Exp. Toxicol. Pathol. 2016, 68, 149–156. [Google Scholar] [CrossRef]
- Alexandrov, K.; Rojas, M.; Satarug, S. The critical DNA damage by benzo(a)pyrene in lung tissues of smokers and approaches to preventing its formation. Toxicol. Lett. 2010, 198, 63–68. [Google Scholar] [CrossRef]
- Wang, S.; Chanock, S.; Tang, D.; Li, Z.; Jedrychowski, W.; Perera, F.P. Assessment of interactions between PAH exposure and genetic polymorphisms on PAH-DNA adducts in African American, Dominican, and Caucasian mothers and newborns. Cancer Epidemiol. Biomark. Prev. 2008, 17, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Li, Z.W. Research progress of the gene polymorphisms of metabolic enzyme related to polycyclic aromatic hydrocarbons risk of preterm birth. Zhonghua Yu Fang Yi Xue Za Zhi 2016, 50, 463–467. [Google Scholar] [CrossRef]
- Gao, M.; Li, Y.; Xue, X.; Long, J.; Chen, L.; Shah, W.; Kong, Y. Impact of AhR, CYP1A1 and GSTM1 genetic polymorphisms on TP53 R273G mutations in individuals exposed to polycyclic aromatic hydrocarbons. Asian Pac. J. Cancer Prev. 2014, 15, 2699–2705. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Lee, Y.S.; Lee, D.H.; Kim, D.S. Polycyclic aromatic hydrocarbons are associated with insulin receptor substrate 2 methylation in adipose tissues of Korean women. Environ. Res. 2016, 150, 47–51. [Google Scholar] [CrossRef]
- Peng, Q.; Chen, H.; Huo, J.R. Alcohol consumption and corresponding factors: A novel perspective on the risk factors of esophageal cancer. Oncol. Lett. 2016, 11, 3231–3239. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Long, J.; Li, Y.; Shah, W.; Fu, L.; Liu, J.; Wang, Y. Mitochondrial decay is involved in BaP-induced cervical damage. Free Radic. Biol. Med. 2010, 49, 1735–1745. [Google Scholar] [CrossRef]
- Gao, M.; Li, Y.; Long, J.; Shah, W.; Fu, L.; Lai, B.; Wang, Y. Induction of oxidative stress and DNA damage in cervix in acute treatment with benzo[a]pyrene. Mutat. Res. 2011, 719, 52–59. [Google Scholar] [CrossRef]
- Harris, K.L.; Banks, L.D.; Mantey, J.A.; Huderson, A.C.; Ramesh, A. Bioaccessibility of polycyclic aromatic hydrocarbons: Relevance to toxicity and carcinogenesis. Expert Opin. Drug Metab. Toxicol. 2013, 9, 1465–1480. [Google Scholar] [CrossRef] [Green Version]
- Rao, P.S.; Kumar, S. Polycyclic aromatic hydrocarbons and cytochrome P450 in HIV pathogenesis. Front. Microbiol. 2015, 6, 550. [Google Scholar] [CrossRef] [Green Version]
- Pankow, J.F.; Watanabe, K.H.; Toccalino, P.L.; Luo, W.; Austin, D.F. Calculated cancer risks for conventional and “potentially reduced exposure product” cigarettes. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 584–592. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat. Rev. Cancer 2003, 3, 733–744. [Google Scholar] [CrossRef]
- Prignot, J.J. Recent contributions of air- and biomarkers to the control of secondhand smoke (SHS): A review. Int. J. Environ. Res. Public Health 2011, 8, 648–682. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem. Res. Toxicol. 1998, 11, 559–603. [Google Scholar] [CrossRef]
- Hoffmann, D.; Hecht, S.S. Nicotine-derived N-nitrosamines and tobacco-related cancer: Current status and future directions. Cancer Res. 1985, 45, 935–944. [Google Scholar]
- Hecht, S.S. DNA adduct formation from tobacco-specific N-nitrosamines. Mutat. Res. 1999, 424, 127–142. [Google Scholar] [CrossRef]
- Hecht, S.S.; Hoffmann, D. Tobacco-specific nitrosamines, an important group of carcinogens in tobacco and tobacco smoke. Carcinogenesis 1988, 9, 875–884. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, D.; Brunnemann, K.D.; Prokopczyk, B.; Djordjevic, M.V. Tobacco-specific N-nitrosamines and Areca-derived N-nitrosamines: Chemistry, biochemistry, carcinogenicity, and relevance to humans. J. Toxicol. Environ. Health 1994, 41, 1–52. [Google Scholar] [CrossRef]
- Maser, E.; Richter, E.; Friebertshäuser, J. The identification of 11 beta-hydroxysteroid dehydrogenase as carbonyl reductase of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Eur. J. Biochem. 1996, 238, 484–489. [Google Scholar] [CrossRef]
- Laag, E.; Majidi, M.; Cekanova, M.; Masi, T.; Takahashi, T.; Schuller, H.M. NNK activates ERK1/2 and CREB/ATF-1 via beta-1-AR and EGFR signaling in human lung adenocarcinoma and small airway epithelial cells. Int. J. Cancer 2006, 119, 1547–1552. [Google Scholar] [CrossRef]
- Hoffmann, D.; Hoffmann, I.; El-Bayoumy, K. The less harmful cigarette: A controversial issue. a tribute to Ernst L. Wynder. Chem. Res. Toxicol. 2001, 14, 767–790. [Google Scholar] [CrossRef]
- Arimoto-Kobayashi, S.; Sakata, H.; Mitsu, K.; Tanoue, H. A possible photosensitizer: Tobacco-specific nitrosamine, 4-(N-methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), induced mutations, DNA strand breaks and oxidative and methylative damage with UVA. Mutat. Res. 2007, 632, 111–120. [Google Scholar] [CrossRef]
- Hang, B.; Sarker, A.H.; Havel, C.; Saha, S.; Hazra, T.K.; Schick, S.; Jacob, P., 3rd; Rehan, V.K.; Chenna, A.; Sharan, D.; et al. Thirdhand smoke causes DNA damage in human cells. Mutagenesis 2013, 28, 381–391. [Google Scholar] [CrossRef]
- Sarker, A.H.; Hang, B. Tobacco-specific nitrosamine 1-(N-methyl-N-nitrosamino)-1-(3-pyridinyl)-4-butanal (NNA) causes DNA damage and impaired replication/transcription in human lung cells. PLoS ONE 2022, 17, e0267839. [Google Scholar] [CrossRef]
- Horton, J.K.; Baker, A.; Berg, B.J.; Sobol, R.W.; Wilson, S.H. Involvement of DNA polymerase beta in protection against the cytotoxicity of oxidative DNA damage. DNA Repair 2002, 1, 317–333. [Google Scholar] [CrossRef]
- Anthony, H.M. Reactive changes in the blood of smokers and the development of arterial diseases and COPD, a review: Evidence of associations between changes and subsequent disease with implications for the evaluation of harmful effects of cigarettes, and for susceptibility to the chronic effects of inhaled pollutants. Rev. Environ. Health 1989, 8, 25–86. [Google Scholar]
- Matta, J.A. Nicotinic acetylcholine receptor redux: Discovery of accessories opens therapeutic vistas. Science 2021, 373, eabg6539. [Google Scholar] [CrossRef]
- Changeux, J.P. Nicotine addiction and nicotinic receptors: Lessons from genetically modified mice. Nat. Rev. Neurosci. 2010, 11, 389–401. [Google Scholar] [CrossRef]
- Gotti, C.; Fornasari, D.; Clementi, F. Human neuronal nicotinic receptors. Prog. Neurobiol. 1997, 53, 199–237. [Google Scholar] [CrossRef]
- Singh, S.; Pillai, S.; Chellappan, S. Nicotinic acetylcholine receptor signaling in tumor growth and metastasis. J. Oncol. 2011, 2011, 456743. [Google Scholar] [CrossRef]
- Gu, S.; Matta, J.A.; Lord, B.; Harrington, A.W.; Sutton, S.W.; Davini, W.B.; Bredt, D.S. Brain α7 Nicotinic Acetylcholine Receptor Assembly Requires NACHO. Neuron 2016, 89, 948–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arredondo, J.; Chernyavsky, A.I.; Grando, S.A. Nicotinic receptors mediate tumorigenic action of tobacco-derived nitrosamines on immortalized oral epithelial cells. Cancer Biol. 2006, 5, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.H.; Tseng, T.Y.; Wu, C.Y.; Chen, P.Y.; Chen, M.F.; Kuo, J.S.; Lee, T.J. Memantine inhibits α3β2-nAChRs-mediated nitrergic neurogenic vasodilation in porcine basilar arteries. PLoS ONE 2012, 7, e40326. [Google Scholar] [CrossRef]
- Jull, B.A.; Plummer, H.K., 3rd; Schuller, H.M. Nicotinic receptor-mediated activation by the tobacco-specific nitrosamine NNK of a Raf-1/MAP kinase pathway, resulting in phosphorylation of c-myc in human small cell lung carcinoma cells and pulmonary neuroendocrine cells. J. Cancer Res. Clin. Oncol. 2001, 127, 707–717. [Google Scholar] [CrossRef]
- Arredondo, J.; Chernyavsky, A.I.; Grando, S.A. The nicotinic receptor antagonists abolish pathobiologic effects of tobacco-derived nitrosamines on BEP2D cells. J. Cancer Res. Clin. Oncol. 2006, 132, 653–663. [Google Scholar] [CrossRef]
- Askari, M.D.; Tsao, M.S.; Schuller, H.M. The tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates proliferation of immortalized human pancreatic duct epithelia through beta-adrenergic transactivation of EGF receptors. J. Cancer Res. Clin. Oncol. 2005, 131, 639–648. [Google Scholar] [CrossRef]
- Schuller, H.M.; Tithof, P.K.; Williams, M.; Plummer, H., 3rd. The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid. Cancer Res. 1999, 59, 4510–4515. [Google Scholar]
- Schuller, H.M.; Al-Wadei, H.A.; Majidi, M. Gamma-aminobutyric acid, a potential tumor suppressor for small airway-derived lung adenocarcinoma. Carcinogenesis 2008, 29, 1979–1985. [Google Scholar] [CrossRef] [Green Version]
- Kawai, H.; Berg, D.K. Nicotinic acetylcholine receptors containing alpha 7 subunits on rat cortical neurons do not undergo long-lasting inactivation even when up-regulated by chronic nicotine exposure. J. Neurochem. 2001, 78, 1367–1378. [Google Scholar] [CrossRef]
- National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health. Reports of the Surgeon General. In The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General; Centers for Disease Control and Prevention (US): Atlanta, GA, USA, 2014. [Google Scholar]
- Geijer, R.M.; Sachs, A.P.; Verheij, T.J.; Salomé, P.L.; Lammers, J.W.; Hoes, A.W. Incidence and determinants of moderate COPD (GOLD II) in male smokers aged 40-65 years: 5-year follow up. Br. J. Gen. Pract. 2006, 56, 656–661. [Google Scholar]
- Gwilt, C.R.; Donnelly, L.E.; Rogers, D.F. The non-neuronal cholinergic system in the airways: An unappreciated regulatory role in pulmonary inflammation? Pharm. Ther. 2007, 115, 208–222. [Google Scholar] [CrossRef]
- Schuller, H.M. Carbon dioxide potentiates the mitogenic effects of nicotine and its carcinogenic derivative, NNK, in normal and neoplastic neuroendocrine lung cells via stimulation of autocrine and protein kinase C-dependent mitogenic pathways. Neurotoxicology 1994, 15, 877–886. [Google Scholar]
- Schuller, H.M.; McGavin, M.D.; Orloff, M.; Riechert, A.; Porter, B. Simultaneous exposure to nicotine and hyperoxia causes tumors in hamsters. Lab. Investig. 1995, 73, 448–456. [Google Scholar]
- Rosa, J.G.; Prokopczyk, B.; Desai, D.H.; Amin, S.G.; El-Bayoumy, K. Elevated 8-hydroxy-2’-deoxyguanosine levels in lung DNA of A/J mice and F344 rats treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and inhibition by dietary 1,4-phenylenebis(methylene)selenocyanate. Carcinogenesis 1998, 19, 1783–1788. [Google Scholar] [CrossRef] [Green Version]
- Chung, F.L.; Xu, Y. Increased 8-oxodeoxyguanosine levels in lung DNA of A/J mice and F344 rats treated with the tobacco-specific nitrosamine 4-(methylnitrosamine)-1-(3-pyridyl)-1-butanone. Carcinogenesis 1992, 13, 1269–1272. [Google Scholar] [CrossRef]
- Bilodeau, J.F.; Wang, M.; Chung, F.L.; Castonguay, A. Effects of nonsteroidal antiinflammatory drugs on oxidative pathways in A/J mice. Free Radic. Biol. Med. 1995, 18, 47–54. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Z.; Lu, T.; Zhang, L.; Cheng, J.; Fu, X.; Hou, Y. Toxic effects of 1-(N-methyl-N-nitrosamino)-1-(3-pyridinyl)-4-butanal on the maturation and subsequent development of murine oocyte. Ecotoxicol. Environ. Saf. 2019, 181, 370–380. [Google Scholar] [CrossRef]
- Hegde, V.; Wang, M.; Deutsch, W.A. Human ribosomal protein S3 interacts with DNA base excision repair proteins hAPE/Ref-1 and hOGG1. Biochemistry 2004, 43, 14211–14217. [Google Scholar] [CrossRef]
- Park, Y.J.; Kim, T.S.; Kim, E.H.; Kim, H.D.; Kim, J. Ribosomal protein S3 is a novel negative regulator of non-homologous end joining repair of DNA double-strand breaks. FASEB J. 2020, 34, 8102–8113. [Google Scholar] [CrossRef] [Green Version]
- Hang, B.L.; Chenna, A. Novel NNA-DNA Adducts as Biomarkers for Detecting Exposure to Thirdhand Smoke. U.S. Patent 10,538,549, 2020. [Google Scholar]
- Molimard, R. The European report “Lifting the Smoke-Screen”: Epidemiological study or manipulation? Rev. Epidemiol. Sante Publique 2008, 56, 286–290. [Google Scholar] [CrossRef]
- Hellström-Lindahl, E.; Nordberg, A. Smoking during pregnancy: A way to transfer the addiction to the next generation? Respiration 2002, 69, 289–293. [Google Scholar] [CrossRef]
- Roberts, J.W.; Dickey, P. Exposure of children to pollutants in house dust and indoor air. Rev. Environ. Contam. Toxicol. 1995, 143, 59–78. [Google Scholar] [CrossRef]
- Martins-Green, M.; Adhami, N.; Frankos, M.; Valdez, M.; Goodwin, B.; Lyubovitsky, J.; Dhall, S.; Garcia, M.; Egiebor, I.; Martinez, B.; et al. Cigarette smoke toxins deposited on surfaces: Implications for human health. PLoS ONE 2014, 9, e86391. [Google Scholar] [CrossRef] [Green Version]
- de Zeeuw, P.; Zwart, F.; Schrama, R.; van Engeland, H.; Durston, S. Prenatal exposure to cigarette smoke or alcohol and cerebellum volume in attention-deficit/hyperactivity disorder and typical development. Transl. Psychiatry 2012, 2, e84. [Google Scholar] [CrossRef]
- Matt, G.E.; Quintana, P.J.; Hovell, M.F.; Bernert, J.T.; Song, S.; Novianti, N.; Juarez, T.; Floro, J.; Gehrman, C.; Garcia, M.; et al. Households contaminated by environmental tobacco smoke: Sources of infant exposures. Tob. Control 2004, 13, 29–37. [Google Scholar] [CrossRef]
- Roels, H.; Hubermont, G.; Buchet, J.P.; Lauwerys, R. Placental transfer of lead, mercury, cadmium, and carbon monoxide in women. III. Factors influencing the accumulation of heavy metals in the placenta and the relationship between metal concentration in the placenta and in maternal and cord blood. Environ. Res. 1978, 16, 236–247. [Google Scholar] [CrossRef]
- Matt, G.E.; Quintana, P.J.E.; Hoh, E.; Dodder, N.G.; Mahabee-Gittens, E.M.; Padilla, S.; Markman, L.; Watanabe, K. Tobacco smoke is a likely source of lead and cadmium in settled house dust. J. Trace Elem. Med. Biol. 2021, 63, 126656. [Google Scholar] [CrossRef]
- Yang, A.M.; Lo, K.; Zheng, T.Z.; Yang, J.L.; Bai, Y.N.; Feng, Y.Q.; Cheng, N.; Liu, S.M. Environmental heavy metals and cardiovascular diseases: Status and future direction. Chronic Dis. Transl. Med. 2020, 6, 251–259. [Google Scholar] [CrossRef]
- Bertone, E.R.; Snyder, L.A.; Moore, A.S. Environmental tobacco smoke and risk of malignant lymphoma in pet cats. Am. J. Epidemiol. 2002, 156, 268–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benowitz, N.L.; Nardone, N.; Jain, S.; Dempsey, D.A.; Addo, N.; St Helen, G.; Jacob, P., 3rd. Comparison of Urine 4-(Methylnitrosamino)-1-(3)Pyridyl-1-Butanol and Cotinine for Assessment of Active and Passive Smoke Exposure in Urban Adolescents. Cancer Epidemiol. Biomark. Prev. 2018, 27, 254–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vardavas, C.I.; Fthenou, E.; Patelarou, E.; Bagkeris, E.; Murphy, S.; Hecht, S.S.; Connolly, G.N.; Chatzi, L.; Kogevinas, M. Exposure to different sources of second-hand smoke during pregnancy and its effect on urinary cotinine and tobacco-specific nitrosamine (NNAL) concentrations. Tob. Control 2013, 22, 194–200. [Google Scholar] [CrossRef]
- Pratt, K. The Effects of Thirdhand Smoke on Biomarkers of Exposure, Inflammation and Oxidative Stress. UCSF. ProQuest ID: Pratt_ucsf_0034M_11144.REDACTED. Merritt ID: Ark:/13030/m53804q4. Available online: https://escholarship.org/uc/item/91g8450d (accessed on 24 April 2022).
- Jacob, P., 3rd; Goniewicz, M.L.; Havel, C.M.; Schick, S.F.; Benowitz, N.L. Nicotelline: A proposed biomarker and environmental tracer for particulate matter derived from tobacco smoke. Chem. Res. Toxicol. 2013, 26, 1615–1631. [Google Scholar] [CrossRef] [Green Version]
- Weschler, C.J.; Nazaroff, W.W. Dermal uptake of organic vapors commonly found in indoor air. Environ. Sci. Technol. 2014, 48, 1230–1237. [Google Scholar] [CrossRef] [Green Version]
- Bekö, G.; Morrison, G.; Weschler, C.J.; Koch, H.M.; Pälmke, C.; Salthammer, T.; Schripp, T. Measurements of dermal uptake of nicotine directly from air and clothing. Indoor Air 2017, 27, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Torres, S.; Merino, C. Biomarkers of Exposure to Secondhand and Thirdhand Tobacco Smoke: Recent Advances and Future Perspectives. Int. J. Environ. Res. Public Health 2018, 15, 2693. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Major Components | Vapor Pressure Range | Representative Compounds in the Components |
|---|---|---|
| Gas-phase inorganic compounds | >13 kPa | CO2, CO, NH3 |
| Very volatile organic compounds(VVOCs) | >7 to 13 kPa | Formaldehyde, acrolein, 1,3-butadiene, acetylaldehyde |
| Volatile organic compounds(VOCs) | 0.01 to 10 kPa | Benzene, styrene, toluene, 2-butanone, N, N-nitrosodimethylamine, N-nitrosopyrrolidine |
| Semi-volatile organic compounds(SVOCs) | 10−2 to 10−8 kPa | Nicotine, N-nitrosonomicotine, 4-(methylnitrosamino), 1-(3-pyridyl)-1-butanone |
| Particulate organic compounds | <10−8 kPa | Benzo[α]pyrene, benzo[β]fluoranthene, benzo[κ]fluoranthene, solanesol |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.-X.; Lau, A.T.Y.; Xu, Y.-M. Indoor Secondary Pollutants Cannot Be Ignored: Third-Hand Smoke. Toxics 2022, 10, 363. https://doi.org/10.3390/toxics10070363
Wu J-X, Lau ATY, Xu Y-M. Indoor Secondary Pollutants Cannot Be Ignored: Third-Hand Smoke. Toxics. 2022; 10(7):363. https://doi.org/10.3390/toxics10070363
Chicago/Turabian StyleWu, Jia-Xun, Andy T. Y. Lau, and Yan-Ming Xu. 2022. "Indoor Secondary Pollutants Cannot Be Ignored: Third-Hand Smoke" Toxics 10, no. 7: 363. https://doi.org/10.3390/toxics10070363
APA StyleWu, J. -X., Lau, A. T. Y., & Xu, Y. -M. (2022). Indoor Secondary Pollutants Cannot Be Ignored: Third-Hand Smoke. Toxics, 10(7), 363. https://doi.org/10.3390/toxics10070363