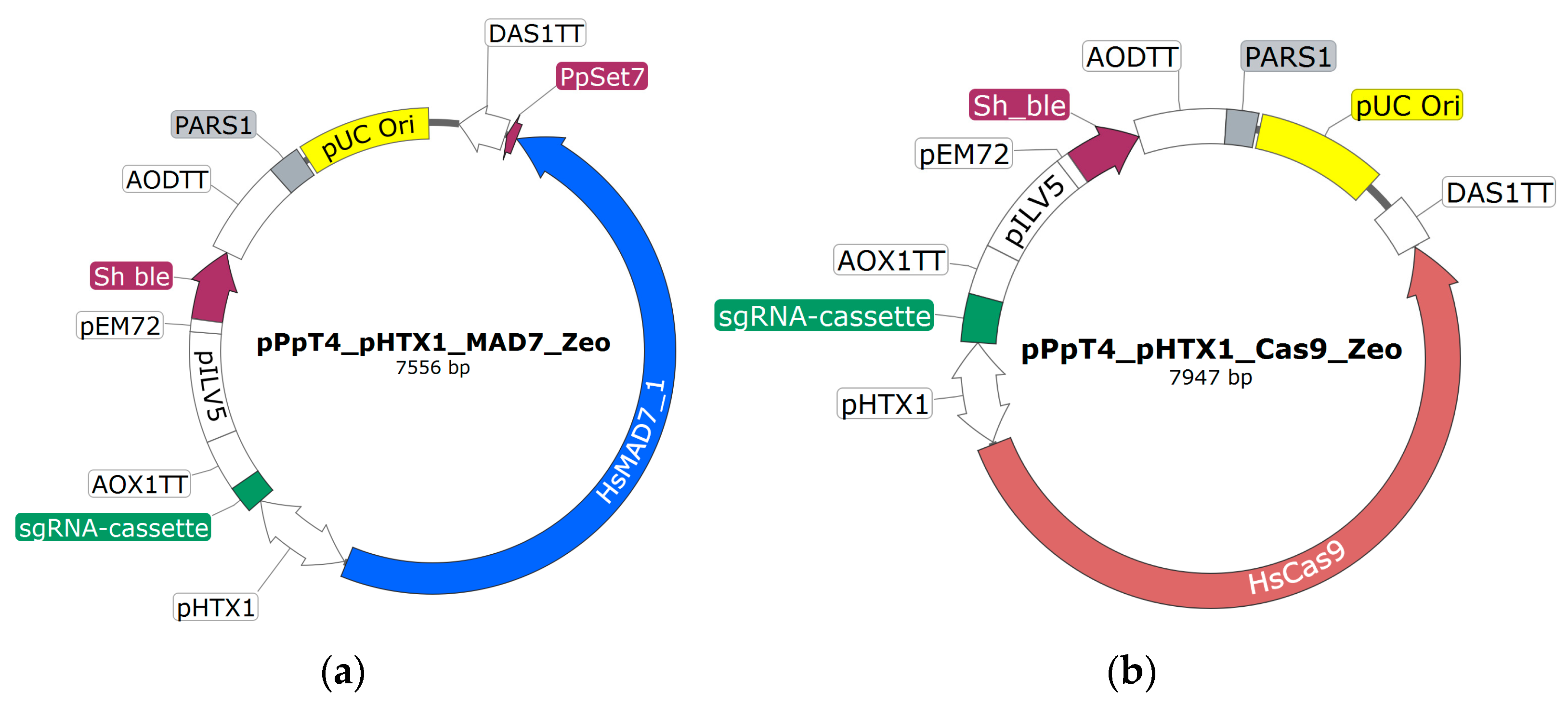
3.1. Setting Up CRISPR-MAD7
Based on the previous experience of establishing a functional CRISPR-Cas9 system in
K. phaffii [
15], it was known that codon optimization of the nuclease, the employed NLS sequence, and the gRNA scaffold sequence are the key to a good working genome-editing tool. Thus, 30 combinations of different
MAD7 codon optimizations, NLS, and gRNA scaffold sequences were tested to find the combination resulting in the best genome-editing efficiency (
Table 3). In total, six different codon-optimized synthetical
MAD7 were ordered. We tested (i) the
E. coli optimized sequence of Inscripta, (ii) a
K. phaffii optimized gene, and (iii) four genes using different codon optimizations for
Homo sapiens. Several NLS were tested: (i) the
S. cerevisiae Setp7 (syn. Rkm4), a ribosomal lysine methyltransferase, homolog in
K. phaffii (
PpSet7); (ii) the Simian Virus 40 large T antigen (SV40, also used by Weninger et al. [
15]); and (iii) the
Xenopus laevis nucleoplasmin sequence (
XlNuc) [
45]. The NLSs were chosen as they should facilitate different translocation levels of the nuclease to the nucleus, as they did for eGFP in a previous study [
45]. Similarly, three distinct gRNA scaffold sequences that facilitate the binding of the nuclease to the gRNA were tested (
Supplementary File S1, S3). The gRNA scaffold sequences were obtained from the website of Incripta and were either found in the FAQ section with apparently no specific assignment to a host organism [
43] or were specifically designated for their use in yeast [
46] and
E. coli [
47], respectively.
To evaluate the performance of the different constructs, we targeted the
GUT1 locus using the same gRNA based on a 5′-TTTA-3′ PAM site (
Supplementary File S1, S7). Since disruption of the
GUT1 results in phenotypes with reduced growth when cultivated on a medium containing glycerol as the sole carbon source (
Figure 2), we could calculate the ratio of slow-growing phenotypes obtained from the randomly selected transformants to which we refer to in the text as disruption efficiency.
The initial studies were performed using the
E. coli and
K. phaffii optimized
MAD7 in combination with the three distinct NLS and gRNA scaffold sequences each (
Table 3, Construct 1–18). Evaluating the efficiency of the different CRISPR-MAD7 constructs showed that the
K. phaffii codon-optimized
MAD7 gene did only work as a functional nuclease when combined with the SV40 and the
E. coli gRNA scaffold sequence (
Table 3, construct 14). All other constructs using the
K. phaffii codon-optimized
MAD7 did not result in slow-growing phenotypes, and hence no genome-editing events occurred (
Table 3). Despite the non-functionality of most tested
K. phaffii optimized
MAD7 constructs, one functional construct still resulted in a gene disruption efficiency of 45%. In the case of the
E. coli optimized
MAD7, the highest gene disruption efficiency (71%) was reached in combination with the
PpSet7 and the
E. coli gRNA scaffold sequence (
Table 3, Construct 8).
To see if the efficiency of the tool could be increased, we tested in the next step the
H. sapiens codon-optimized MAD7 nucleases in combination with different NLS. This was completed because it was shown previously that the
H. sapiens optimized Cas9 worked best in
K. phaffii [
15]. To reduce the number of variables, and since based on the results obtained in this study the
E. coli gRNA scaffold sequence worked best (
Table 3), we decided to only use this gRNA scaffold sequence for testing the
H. sapiens optimized
MAD7 genes. Assessing the functionality of the 12 constructs (
Table 1, Construct 19–30) suggested that
H. sapiens optimized
MAD7 genes work better than the
E. coli and
K. phaffii codon optimization of
MAD7, as target disruption efficiencies of up to 90% were reached (
Table 3, Construct 21). However, like before, large differences between the codon usages and the NLS sequences employed were observed. It appears that
H. sapiens codon usage 1 works best, as all constructs resulted in
GUT1 disruption. Still, we observed a large variation in the disruption efficiency depending on the NLS we used (3%
XlNuc vs. 90%
PpSET). The
MAD7 genes optimized according to
H. sapiens codon usage 2, 3, and 4 did not result in target disruption in combination with the SV40 but seemed to work in combination with the
PpSET7 (
Table 3). Nonetheless, they do not reach the 90%
GUT1 disruption efficiency and were thus not considered for further studies.
Summing up, the most efficient CRISPR-MAD7 system for application in
K. phaffii was obtained by combining the
MAD7 gene optimized according to
H. sapiens codon usage 1 (
Table 3) with the
PpSET7 NLS and the
E. coli gRNA scaffold sequence (
Table 3, Construct 21). This construct resulted in a
GUT1 disruption efficiency of ~90% which was close to the previously reported CRISPR-Cas9 system for
K. phaffii [
15]. Moreover, our data allow for speculations that the key to the efficiency of construct 21 is low transcript levels and low levels of nucleases in the nucleus. This assumption is based on the finding that constructs with
MAD7 optimized according to the distantly related codon usages, which should result due to the codon bias in lower transcript levels [
48], work better than the
K. phaffii codon usage. This is in line with the observed fewer
K. phaffii transformants when the plasmid carrying the
K. phaffii optimized
MAD7 was used for transformation (
Table 3, Construct 10–18), suggesting there is stress and potential lethality caused by off-targeting due to increasing the amounts of nuclease [
49,
50].
3.2. 5′-TTTN-3′ PAM Sites Are Favored in K. phaffii
One of the critical elements when designing gRNA is choosing the correct PAM site. In the case of MAD7, the consensus sequence of the PAM site is 5′-YTTN-3′ in which “Y” can be either a “T” or “C”, and “N” can be one of the four standard nucleotides. To see which PAM site works best for genome engineering using MAD7 in
K. phaffii, gRNAs based on all eight possible PAM sequences were tested (
Table 4). To investigate the potential influence of the gRNA binding site on disruption efficiency, two gRNAs were tested per PAM site sequence (
Table 4). Identical to the previous experiment, we targeted the
GUT1, as gene disruptions can be easily monitored (
Figure 2). Although Construct 21 works best for
GUT1 disruption, we decided to use construct 8 (
Table 3) for assessing the influence of the PAM site, as the disruption rate of 71% allows for the detection of higher and lower target disruption efficiencies.
Our results indicate an influence of the PAM site on
GUT1 disruption efficiency. It appears that the 5′-“Y” in the PAM sequence needs to be occupied by a “T” in
K. phaffii as the constructs based on “C” did not work for gene disruption (
Table 4, #9–16). When looking at the 3′-“N”, our data indicate differences between the PAM sites and might even show that PAM sites can improve the overall efficiency of the CRISPR-MAD7 tool, but due to the experimental set-up (no biological replicates), we cannot say this for certain (
Table 4). Still, we decided to stick to 5′-TTTN-3′ PAMs for subsequent studies, as they seem to work best for CRISPR-MAD7 in
K. phaffii.
Besides the influence of the PAM site sequence on the gene-editing efficiency, we can confirm previous observations [
51,
52] that the gRNAs design is critical for gene disruption, as in two cases only one of the two tested gRNAs per PAM site sequence was functional (
Table 4).
Of note, although the gRNA for targeting the
GUT1 locus in the initial study to set up the CRISPR-MAD7 system (
Table 3) is based on a 5′-TTTA-3′ PAM, the gRNA was different from #1 and #2 in
Table 4. Thus, the difference in target disruption efficiency (79% vs. 21%) is not caused by experimental flaws but supports our point that the gRNA is of major relevance for the efficiency of the CRISPR-MAD7 system. Additionally, further factors like the intra-genetic gRNA target locus, the accessibility of the target sequence, and the position-specific sequence features of the target nucleic acid may also impact the efficiency, as outlined previously [
52,
53]. Therefore, it is suggested that it would be helpful to design and test multiple gRNAs per target gene and to screen a large and diverse set of transformants to increase the chances of successful gene editing.
3.3. Using CRISPR-MAD7 to Edit Other Targets Than the GUT1 Gene
The
GUT1 gene is a convenient target for testing and establishing the CRISPR-MAD7 system in
K. phaffii, since it is easy to screen for gene disruption events and the locus seems to be easily accessible for genome engineering procedures. Nonetheless, to verify the system’s robustness and broader applicability, additional integrated genes in the
K. phaffii genome were targeted and aimed to be disrupted by the established CRISPR-MAD7 tool (
Table 3, Construct 21).
As a next gene, the triosephosphate isomerase (
TPI1) gene was targeted, a central enzyme in glycolysis, that is responsible for the interconversion of dihydroxyacetone phosphate (DHAP) to D-glyceraldehyde 3-phosphate (GAP). Due to its central role in carbon source metabolism, it can on the one hand be considered as a challenging disruption target, because
TPI1 deficient
S. cerevisiae strains show poor growth on most carbon sources, which means there would be a selection for functional Tpi1p on transformation plates [
54,
55], and, on the other hand, it could be used as an interesting knockout target due to the potential use of
TPI1 as selection marker in a
TPI1 deficient host strain [
56,
57]. In addition, several other research teams reported before that such a knock out was not possible before in
K. phaffii (personal communication). Successful
TPI1 inactivation would result in slow growth on glucose and no growth on glycerol as a carbon source and has thus considerable impact on the cell metabolism [
54,
57]. To edit the
TPI1 using the CRISPR-MAD7 system, we designed and tested four distinct gRNAs (
Supplementary File S1, S7) to increase the chances of a successful CRISPR-MAD7 construct. gRNAs 1-3 led to potential
K. phaffii ∆
tpi1 strains. The potential knockout mutants were identified, as they showed, in line with the premise that
TPI1 is essential for glycerol utilization [
57], no growth on glycerol as the sole carbon source. To investigate if indeed the inactivation of
TPI1 is responsible for the inability of these strains to utilize glycerol, the
TPI1 gene was PCR amplified and sequenced. The genotyping showed, as expected, frame-shifting deletions, confirming successful
TPI1 inactivation with CRISPR-MAD7.
The second alternative
K. phaffii target was the
OCH1 gene
. OCH1 encodes for α-1,6-mannosyltransferase, which catalyzes the first step of the hypermannosylation of proteins in the Golgi apparatus in
K. phaffii (and other yeasts) and is, therefore, critical for the glycosylation levels of secretory proteins [
58,
59]. Thus, the inactivation of
OCH1 results in the reduction of mannosylation levels, easing downstream processing and increasing the biopharmaceutical properties of secreted proteins ([
60,
61]) which makes it a highly appreciated target. However,
OCH1 is known from previous genome-engineering efforts as a target with low disruption efficiencies [
58], making it a challenging locus for testing the established CRISPR-MAD7 system. As for the other targets, we were able to inactivate
OCH1 using the MAD7-based CRISPR tool. Still, in the case of
OCH1, we could only observe the characteristic
OCH1 phenotype featuring reduced cell wall integrity and growth as outlined by Krainer et al. [
58], when gRNA 1 (
Supplementary File S1, S7) was used to direct the MAD7 nuclease to the
OCH1 locus highlighting again the importance of the gRNA. The disruption of the
OCH1 reading frame was subsequently confirmed by sequencing the PCR-amplified
OCH1 coding genomic fragment.
Another challenging target for testing the CRISPR-MAD7 system was found in
PMT2. In fungi, protein-O-mannosyltransferases (PMTs) are responsible for protein-O-mannosylation in the ER, and
PMT2 is the highest expressed representative [
62]. While a knockout of the
PMT2 seems to be lethal for
Schizosaccharomyces pombe and
Candida albicans, it was reported in
K. phaffii to lead to the secretion of proteins with a reduced O-glycan site occupancy and O-glycan chain length [
62]. This suggested that the
PMT2 is a difficult but very valuable knockout target, as it brings similar benefits for the secretion of proteins like a
OCH1 knockout [
62,
63,
64]. Like for
OCH1, only one of the six gRNA (gRNA 6,
Supplementary File S1, S7) resulted in editing of the
PMT2 using the CRISPR-MAD7 tool. The multiple potential ∆
pmt2 mutants were identified using their slow-growing phenotype as previously described [
62]. However, when we evaluated the sequencing results to confirm the
PMT2 knockout, we discovered that the slow-growing phenotypes had short frame-shifting indels as well as very large frame-shifting (>1000 bp) and in-frame deletions (363 bp) meaning such deletions were sufficient to alter the Pmt2p activity enough to result in phenotypical characteristics. This supports the observations that
PMT2 is of high importance for
K. phaffii or might be connected to the nuclease itself as discussed in the following section.
At this point, it has to be mentioned that, although we could knockout the TPI, OCH1 and PMT2 using the CRISPR-MAD7 system, the gene disruption efficiency was low, most likely caused by the importance of genes for cellular integrity. Indeed, without pre-screening for the anticipated phenotypical characteristics, such as slow growth, and testing a variety of gRNAs it would be difficult to identify respective knockout strains among other colonies of non-modified cells. Thus, we thought it was misleading to calculate the target disruption efficiencies of the MAD7 system based on those challenging targets.
Thus, we decided to use a previously made
K. phaffii strain, expressing the red fluorescence protein (
DsRed) as the reporter protein, and carrying a Zeocin (Sh ble) resistance cassette for selection, as an alternative knockout target [
37] to the
GUT1 gene. Like in previous experiments, two gRNAs per target were designed and tested (
Supplementary File S1, S7), and the knockout candidates were picked randomly without pre-screening. To detect successful disruption of
DsRed and Sh ble, the obtained clones were screened for their fluorescence levels and their ability to grow on media containing Zeocin, respectively. In accordance with our previous observation, we saw differences between the gRNAs employed for directing the nuclease to the target. In the experiments where
DsRed was the knockout target, 32 out of 42 (76%) screened clones targeted by gRNA 1 and 30 out of 42 (71%), targeted by gRNA 2, showed wildtype-like fluorescence levels suggesting that the
DsRed gene was disrupted (
Table 5). Similar numbers were obtained when Sh ble was chosen as the knockout target, as 29 out of 42 (69%) clones targeted with gRNA 1 and 32 out of 42 (76%) screened clones targeted with gRNA 2 lost their ability to grow on Zeocin supplemented media (
Table 5). This confirms that the CRISPR-MAD7 system is applicable for efficient ORF disruptions of native and non-native genes in
K. phaffii.
3.4. Systematic Evaluation of the Genome-Wide-Editing Efficiency of the CRISPR-Cas9 and the CRISPR-MAD7 Systems in K. phaffii
Having access to a CRISPR-MAD7 system (
Table 3, Construct 21), which showed the high gene-editing performance in
K. phaffii for targeting various genes and previously Weninger et al. developed, by now the well-established, CRISPR-Cas9 tool [
15], a comparison of the two systems as tools for high-throughput knock-out generation was initiated. For this purpose, 259 genes encoding kinases were selected, as it was previously shown that some kinases can be successfully disrupted in
K. phaffii using classical genome engineering techniques [
65], although a significant number of those regulatory proteins can be assumed to be essential for
K. phaffii. Furthermore, the high number of target genes decreased the bias that comes from gRNAs’ design or the position of individual genes at chromosomes. Therefore, it enabled determination of a genome-wide-editing efficiency, which can be applied to most of the genes of both systems.
Since Cas9 and MAD7 require different PAM sites, two distinct sets of target specific gRNAs (
Supplementary File S2, Table S2) had to be designed for each CRISPR system. 5′-NGG-3′ and 5′-TTTN-3′ were used as consensus sequences of PAM sites Cas9 and MAD7, respectively.
Although in the experiments targeting GUT1, DsRed, and Sh ble genes the disruption efficiency was defined as the ratio of the clones with edited genes to the total number of screened clones in which the same genes were targeted, the different approaches for defining the gene-editing efficiency were used in the comparison study. As we searched for a parameter that characterizes the average efficacy of two CRISPR systems, the genome-wide-editing efficiency was calculated as the ratio of the number of edited genes to the total number of targeted genes.
In total, 181 of the 259 (70%) kinase genes were successfully mutated. In total, 169 of 259 (~65%) targeted kinases were edited when the CRISPR-Cas9 system was used, and 59 of 259 (~23%) genes were edited by employing the CRISPR-MAD7 system (
Table 6,
Supplementary File S2, Table S6, Supplementary File S3). A total of 47 of the kinases were mutated using both tools. Consequently, 122 genes could only be mutated using Cas9, and 12 only using MAD7. Thus, the data indicated that kinase open reading frames can be successfully engineered using both CRISPR methods, but the genome-wide-editing efficiency of the CRISPR-Cas9 was approximately three times higher than the genome-wide-editing efficiency of the employed CRISPR-MAD7 system.
3.6. Identification of Favorable PAM Sequences for the CRISPR-MAD7and the CRISPR-Cas9 Systems in K. phaffii
The preliminary testing of the CRISPR-MAD7 system indicated that the PAM site used for gRNA design might affect the target disruption efficiency of the MAD7 endonuclease but did not show a clear trend (
Table 3). In addition, it was recently reported that the PAM site can influence the cleavage efficacy of the CRISPR-Cas9 system [
67]. Thus, we performed some simple calculations to test the influence of the PAM sites in a larger setup and to identify/confirm preferable PAM sites for both systems.
For the calculations, the gRNAs used in the CRISPR-MAD7 system were classified into four groups according to the PAM sequences. In our study, the least-selected PAM site sequences from the CCTOP tool were 5′-TTTA-3′ (14%) and 5′-TTTT-3′ (21%), whereas the other two PAM sites (5′-TTTG-3′ and 5′-TTTC-3′) were chosen from the tool with similar probability (~34% and ~31%, respectively). Subsequently, the absolute and relative numbers of gRNAs and editing events were calculated for each group. The gRNAs which were designed based on the 5′-TTTC-3′ and 5′-TTTA-3′ PAM-site sequences resulted in editing efficiency rates of 28% and 27%, respectively. The 5′-TTTT-3′ and 5′-TTTG-3′ PAM-site sequences had editing efficiencies of 19%, and were slightly less efficient but still very comparable (
Table 7). Thus, we could confirm the trend observed in
Table 4 suggesting that all four 5′-TTTN-3′ based PAM sites are suitable to obtain gene knockouts in
K. phaffii using the CRISPR-MAD7 tool.
Similarly, as for the MAD7-based system, the gRNAs used in the CRISPR-Cas9 system were grouped according to the 5′-NGG-3′ PAM consensus sequences into four classes. In this case, the 5′-AGG-3′ (32%) and 5′-TGG-3′ (40%) based gRNAs occurred more frequently than the 5′-CGG-3′ (15%) and 5′-GGG-3′ (13%) based gRNAs. Consequently, the 5′-AGG-3′ and 5′-TGG-3′ based gRNAs were involved in the majority of the observed editing events (
Table 8). However, the calculation of the average gene-editing efficiency for each gRNA revealed that using 5′-CGG-3′ and 5′-GGG-3′ based gRNAs result in ~76% of cases in an editing event. In contrast, 5′-AGG-3′ and 5′-TGG-3′ based gRNAs appear to be less efficient, as the gene-editing efficiencies observed were 61 and 62%, respectively (
Table 8). Thus, the obtained data suggested that “C” and “G” were the preferred occupations for “N” in the 5′-NGG-3′ PAM consensus sequences in
K. phaffii for the CRISPR-Cas9 system, but, like for the CRISPR-MAD7, the differences in editing efficiency observed between the used PAM sites is not large and will most likely not be the key for an gRNA to be functional or not.
3.7. Comparison of HDR-Mediated Gene Editing of CRISPR-MAD7 and CRISPR-Cas9
The obvious contradiction between the results obtained during the initial establishment and the genome-wide testing of the CRISPR-MAD7 platform needed further investigation. Although the differences in sample sizes may be a possible explanation for the contradiction, another obvious hypothesis was that the staggered ends resulting from the MAD7 cutting mechanism might be less prone to errors when microhomologies are present or repaired using a simple ligase process rather than a non-homologous end-joining process of blunt-ended fragments after double-strand breaks caused by the Cas9 nuclease. If true, this phenomenon could be masking the actual genome-wide-editing efficiency of the CRISPR-MAD7 system. Despite the fact that microhomology-mediated end joining (MMEJ) is considered an error-prone repair mechanism in the literature [
68,
69], investigation of the above-mentioned hypothesis seemed worthwhile.
In order to investigate this hypothesis, it was decided to try to detect DSB events, if they indeed take place, that might be triggered by CRISPR-MAD7 plasmids which did not provide indels in the genome-wide-editing study. However, this time, the occurrence of DSB was checked not by looking for indels caused by NHEJ repair but by seeking knock-in events provided by HDR. For that reason, in addition to BSYBG10, the BSYBG10 Δ
ku70 strain (bisy GmbH, Hofstaetten an der Raab, Austria) was used in this experiment. This strain is known for its significantly reduced ability to repair DSB using NHEJ and due to the fact it favors HDR with donor DNA [
14].
First of all, seven CRISPR-MAD7 plasmids targeting different genes and, based on previous experiments, that were considered non-functional (not able to cause indels) were selected. In parallel, seven functional CRISPR-Cas9 plasmids for the same genes were picked as positive controls which proved that knock-in events are possible in the chosen target genes. Both sets of plasmids and the corresponding donor DNA fragments were used for co-transformation of the
K. phaffii strains BSYBG10 and BSYBG10 Δ
ku70. After that, between four and seven clones from each transformation were analyzed to detect the occurrence of NHEJ- or HDR-mediated editing events. Results obtained during this experiment are presented in
Table 9 under the subtitle “Main group” and they indicate that the CRISPR-MAD7 plasmids whose usage did not lead to indels’ generation are also not able to edit genes through HDR (0/7). As for the CRISPR-Cas9 plasmids, they successfully provided HDR integration events in BSYBG10 Δ
ku70 (7/7), though relatively short homologous arms and a low donor template concentration were used [
70]. Since the MAD7 system had never been used for HDR-mediated integration, an additional control group including functional CRISPR-MAD7 and CRISPR-Cas9 plasmids was necessary.
For the control group, nine genes and, corresponding with them, functional CRISPR-MAD7 and CRISPR-Cas9 plasmids were selected. As is the case with the main group, BSYBG10 and BSYBG10 Δ
ku70 were transformed with CRISPR plasmids, and the donor DNA fragments, and the same analysis was completed. The summary of the results are presented in
Table 9 under the subtitle “Control group”. Although the functional CRISPR-MAD7 plasmids indeed generate indels in BSYBG10 (8/9), their efficiency in HDR-mediated integration in the Δ
ku70 strain is noticeably lower (4/9). This phenomenon points out that the CRISPR-MAD7 system might be sensitive to some experimental conditions such as the concentration of donor DNA [
70]. In contrast, the CRISPR-Cas9 demonstrated high performance under the current experimental conditions, and small fluctuations in HDR-mediated editing in Δ
ku70 (7/9) can be caused by the small number of screened clones.
To sum up, the experiment empirical evidence seriously undermined the initial hypothesis linking the staggering ends with a high chance of errorless DSB repair, but still enabling relatively efficient repair and gene editing using HDR using donor DNA. Despite the fact that the sample size and the number of analyzed transformants was quite limited, these data confirm the objectivity of the previously established genome-wide-editing efficiency of the CRISPR-MAD7 platform. In addition, the advantage of
ku70 deletion strains for homologous recombination was confirmed, as well as their disadvantage for mutagenesis using error-prone NHEJ repair. Additional information including the number of edited clones for each individual targeted gene is available in
Table S7 (Supplementary File S2).


 ,
, 
{kind=link}
{kind=link}

