1. Introduction
Micro- and meso-porous carbons are widely employed in a lot of applications ranging from molecular sieve and gas storage up to catalysis [
1,
2]. To optimize the sorption capacity of carbons, the morphological and textural features must be suitably designed [
3,
4]. Indeed, a tailored porosity is often necessary for specific applications such as hydrogen storage requiring a high value of micropore volume to achieve large hydrogen storage capacity at room temperature [
3,
5].
Carbon materials are lighter than inorganic compounds, which represents an advantage for storage systems [
6]. Most of carbons are obtained as powders, which limits their applicability when macroscopic morphologies are required [
2]. Nevertheless, activated carbon in a structured form is not attainable due to the difficult cohesion of carbon powder particles requiring the use of ligands. Lozano-Castelló et al. [
7] investigated a quite large number of binders to prepare cylindrical shaped activated carbons and found that, although all binders provide good mechanical properties, the adsorption capacity of the activated carbon monolith was reduced compared to that of the starting carbon material due to the partial blocking of porosity. The extent of micropore blocking depends on the type of binder. Commercially activated carbon monoliths are also available, consisting of a carbon layer on a ceramic substrate with a limited adhesion and a loading of about 50%. The activated carbon layer necessarily contains some binder.
For this reason, the template synthesis of porous carbon using a replica technique has attracted a lot of interest in the last two decades [
8,
9], in particular nanocasting of carbon replica on siliceous porous materials. The application of this technique allows a proper modulation of textural properties, tailored to the final use [
3]. According to this method, silica with interconnected porosity is impregnated with a carbon precursor, which is then pyrolyzed in order to obtain the activated carbon. After dissolution of the silica framework, a porous ordered carbon foam is obtained representing the negative replica of the starting template [
8,
9].
Nevertheless, the hard-templating method involves a quite complex preparation procedure including the preparation of the nanostructured silica template, the impregnation with the carbon precursor, the pyrolysis of the precursor and the etching of the templates with HF solution [
8]. To avoid the necessity for several washing treatments after the etching step, Zhang et al. [
9] proposed NaOH etching of the carbon−silica templates followed by filtration and drying, thus obtaining an alkaline mesoporous carbon with high performance in H
2S adsorption at room temperature, due to the introduction of basic properties greatly enhancing the sorption capacity of the acid.
One of the most common carbon precursors, furfuryl alcohol (FA), is generally introduced into silica pores together with oxalic acid which represents the polymerization catalyst [
8]. Polymerization of FA is also activated by mineral acids (H
2SO
4, etc.), organic acid (
p-toluenesulphonic, etc.), acid zeolites (HY, HZSM5) and Lewis acid (I
2, SnCl
4, TiCl
4) [
10]. Cesano et al. [
10] proposed for the first time ZnCl
2 as an acid promoter for FA polymerization at 60–70 °C to obtain porous ZnO–carbon composites characterized by a uniform layer of highly dispersed ZnO crystallites on the surface of the carbon matrix or, at high pyrolysis temperatures, of a pure carbon phase containing holes whose distribution and size is driven by the Zn content.
Wei et al. [
11] also studied ZnO-porous carbon composites starting from FA/ZnCl
2/water solution used to dip-coat glass mats focusing on the effect of the pyrolysis time at 450 °C on the morphology of ZnO crystals more than on that of the carbon matrix.
It has been reported that the introduction of transition metals or metal oxides particles into carbon nanotubes enhances the hydrogen storage capacity [
12]. Furthermore, the addition of gold, nickel, copper, or palladium strongly increases the electrochemical storage of H
2 on carbons [
13].
The good promoting effect of both zinc and copper finely dispersed on activated carbon granular particles at room temperature was recently also demonstrated for H
2S adsorption [
14,
15,
16].
The work of Cesano et al. [
10,
17] suggested the possibility of dispersing the metal promoters directly during the synthesis of activated carbon, producing a material that could potentially be used in gas storage.
On the basis of this consideration, in this paper, we explored the possibility of also introducing another metal, i.e., copper, that can potentially act as a Lewis acid activator for polymerization of furfuryl alcohol in order to produce activated carbons with special adsorption features determined not only by the textural properties, but also by the presence of a given metal, by suitably tuning the polymerization conditions. ZnCl2 was also investigated as a promoter of FA polymerization, in order to have a direct comparison with the Cu-activated material.
Furthermore, a simple preparation method, avoiding the common complex and long multi-step procedure generally used to produce activated carbon as foam monoliths, is investigated. In other words, this paper provides a methodology to produce activated carbons with a tailored porosity and metal load, required by the specific adsorption application, as foamy monoliths potentially usable for high pressure processes such as gas storage.
3. Results and Discussion
Zn/AC sample. A Zn/AC sample was prepared stirring the FA-ZnCl
2 water solution at different temperatures. The mixture required several days to reach a highly viscous texture at room temperature. Cesano et al. [
17] reported that, at room temperature, a time >12 h was necessary to obtain PFA, whereas in our experiment at room temperature a highly viscous material was obtained after times as long as 10 days.
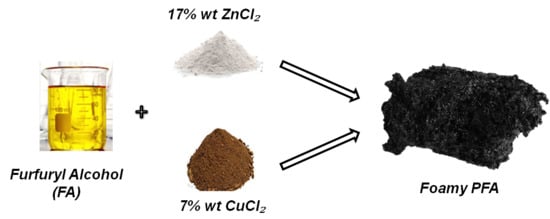
For this reason, another synthesis keeping the same composition of the mixture was performed, increasing the temperature to 80 °C. Under this condition, after about 40 min, an instantaneous formation of a foamy polymer occurred, as shown in
Figure 1. This was considered a positive event. Actually, this could represent an easy and rapid method for producing a structured activated carbon.
Cu/AC sample. Cu
2+ is a stronger Lewis acid than Zn
2+ because of its smaller ionic radius and the higher Pauling electronegativity. As a consequence, a stronger activation of branching and cross-linking can be expected. For this reason, a lower CuCl
2 concentration (7 wt%) was chosen in order to balance the stronger Lewis acid strength of copper chloride with respect to zinc chloride. Indeed, although an absolute classification of metal Lewis acids is not possible because the acidity value strongly depends on the reaction, Kobayashi et al. [
23] classified the Lewis acidity of many metal chlorides on the basis of yield and selectivity in the addition reaction of silyl enolate to an aldehyde and an aldimine. They found 60% yield for CuCl
2 and 23% yield for ZnCl
2. The ratios between the concentration of the two metal chlorides we used was the same as these yields.
Nevertheless, despite the reduction of copper chloride concentration with respect to that of the zinc salt, FA polymerization took place with the formation of the polymer at room temperature in only two hours, compared to the several days required for Zn-activated polymerization at the same temperature, coupled to a violent and uncontrolled formation of carbon foam. The same occurred at 80 °C in only a few minutes. For this reason, FA was polymerized at 0 °C, a condition that allowed a mild and controlled polymerization that occurred in 3 days.
These results show that the temperature of polymerization strongly influences the reaction time, and that if the cation is a stronger Lewis acid the temperature necessary to obtain the polymerization in a given time shifts to lower values. Of course, experimental conditions must be carefully chosen to carry out a controlled formation of foamy PFA.
The first explanation of the phenomenon of rapid polymerization of the FA was given by Dunlop and Peters [
24]. They proposed a scheme of two simultaneous reactions occurring during the polymerization of FA in acid aqueous solutions, both involving the condensation of the OH groups. Then, branching and cross-linking can take place, and the rate at which these reactions occur increases with the formation of oligomers. By increasing branching and cross-linking, the liquid solution become more viscous and darker. The formation of foamy polymers goes through a gelly phase. Vaporization of water produced from branching and cross-linking due to the exothermal character of these reaction likely creates the cavities of the PFA foam.
It must be noted that, under our conditions, the presence of water different from that produced by the polymerization reactions, introduced from the very beginning into FA for the dissolution of the metal chlorides, can contribute to the formation of a foamy material, rapidly evaporating when the exothermal reactions take place.
Due to the too long time required to polymerize at room temperature for the Zn-activated system and to the uncontrolled polymerization of the Cu-activated system at 25 and 80 °C, characterization was done on PFA obtained at 80 and 0 °C for ZnCl2 and CuCl2 activation, respectively.
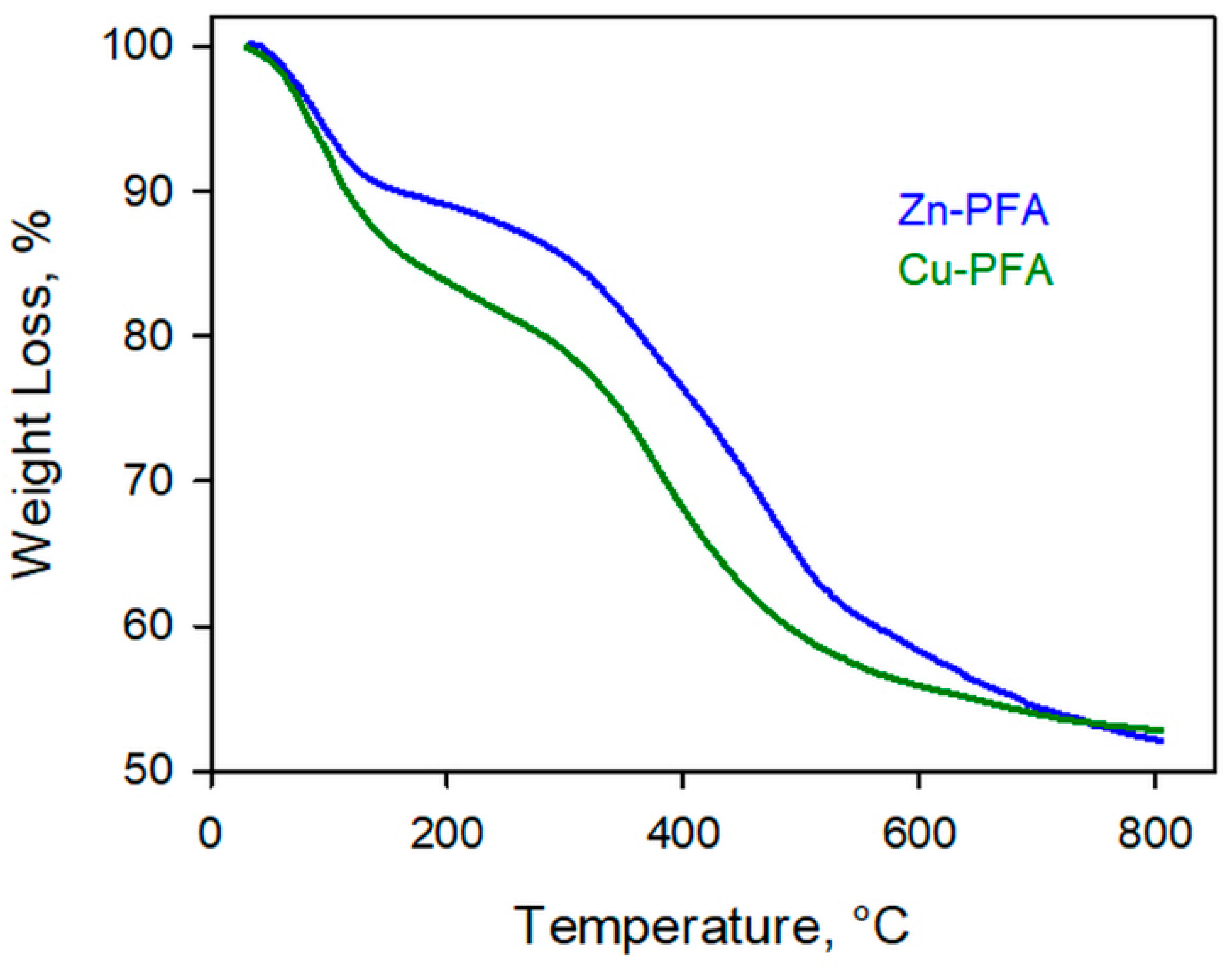
In
Figure 2 the results of TG analysis carried out on both Zn- and Cu-PFA under N
2 are reported.
Both samples undergo two main weight losses. The first, at T < 150 °C, was attributed to the sample dehydration, whereas the second, in the range 350–550 °C, was attributed to PFA degradation leading to the formation of carbon [
25,
26].
On the basis of these results, 600 °C was considered a temperature sufficient to pyrolyze PFA. Nevertheless, some samples were also pyrolyzed at 850 °C to investigate the effect of the temperature on the final properties of the activated carbon. As reported in the Experimental section, this final step was carried out under pure helium flow or under 1000 ppm O
2/He mixture, as proposed by Cesano et al. [
10].
In
Table 1, a list of all samples is reported, with temperature of pyrolysis and composition of pyrolysis gas. The samples are labelled as Me/AC-T, where Me represents the metal, AC the activated carbon and T the temperature of pyrolysis. An asterisk, when present, indicates the presence of O
2 in the pyrolysis gas.
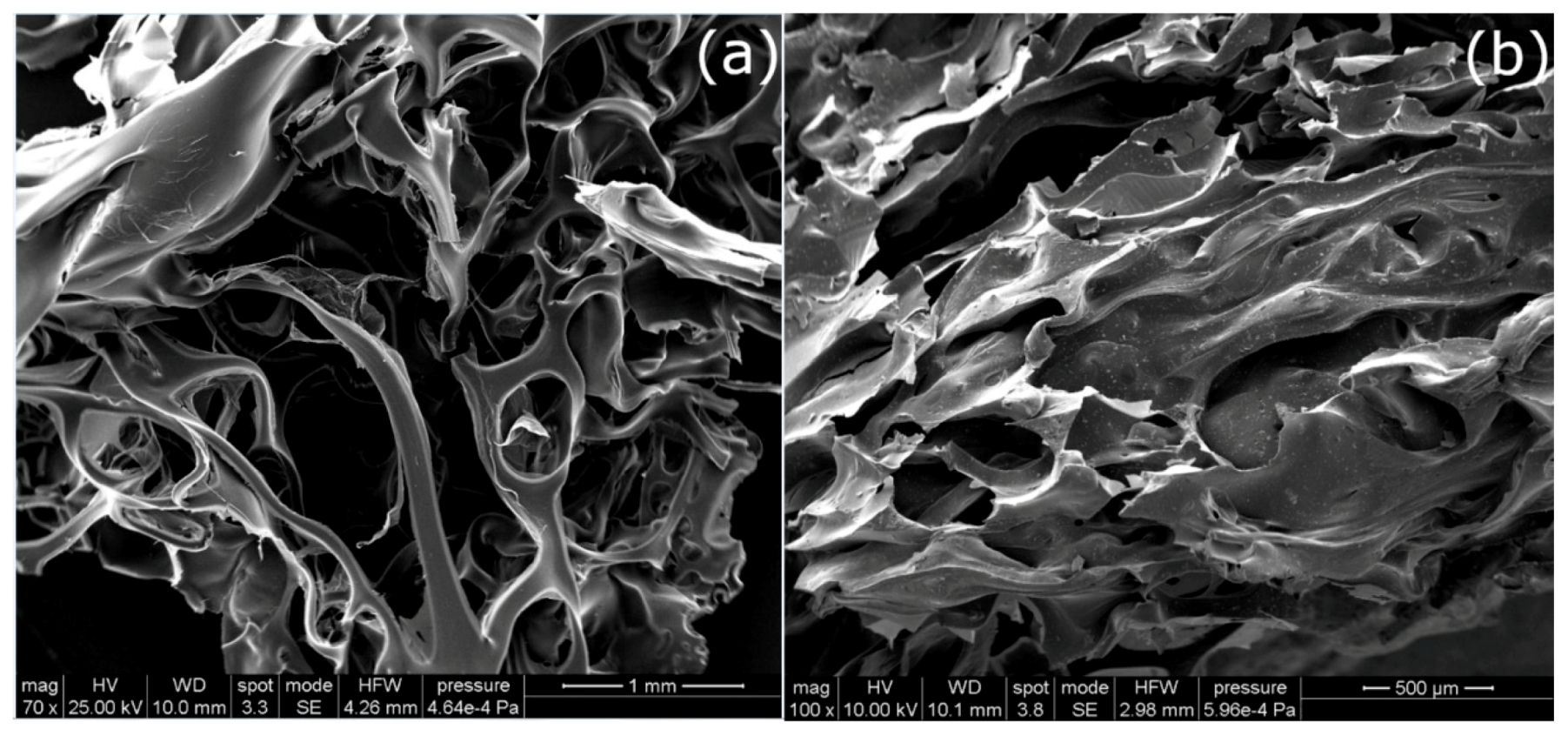
In
Figure 3a the SEM image of Zn/AC-850 is shown in which the foamy structure of AC is easily visible. A similar structure, even if less “open”, was also observed for Cu/AC-850 (
Figure 3b).
A greater magnification highlights the presence of spherical particles on the carbon surface for both Cu/AC-850 and Cu/Ac-600 (
Figure 4b), whereas needle-like particles, observed for Zn/AC-600 sample (
Figure 4a) disappeared when the pyrolysis was carried out at 850 °C for the Zn-activated sample.
The coupled EDX analysis confirmed that the particles detected were made of copper and zinc, respectively.
Results of ICP-MS analysis, carried out for all samples in various places of the foams to determine the actual metal content, are reported in
Table 2. The values reported in the Table represent average contents, which changed by 13 and 15% for copper and zinc, respectively, when moving from one sample portion to another. In the same table, the nominal metal content is also reported (first column) in order to evaluate the fraction of metal preserved upon the thermal treatment. For Zn-based materials, all values were much lower than the nominal one. On the contrary, copper load was quite close to the nominal one, although this sample was treated at 850 °C. This result was expected for the Zn-activated PFA pyrolyzed at 850 °C due to the lower temperature of evaporation of metallic Zn with respect to metallic Cu leading to the complete disappearance of the metal in both Zn samples. This is in agreement with results reported by Cesano et al. [
10] who found by XANES experiments that Zn concentration decreased of about 50% in the samples treated at 600 °C and about 90% in the samples treated at 800 °C with respect to the supposed initial concentration in the PFA. The authors assumed that reduction of Zn(II) to metallic Zn occurred in the temperature range 400–800 °C that easily evaporates due to the high volatility. The evaporation of metallic zinc at T > 800 °C, also if starting from its oxide, was also found by other authors [
27,
28]. Nevertheless, the very low Zn concentration, also for the sample treated at 600 °C, can probably be explained with some confinement on the outer surface of the metal during the fast PFA foam formation. This external portion of PFA foam was, however, removed in all cases without undergoing any subsequent pyrolysis.
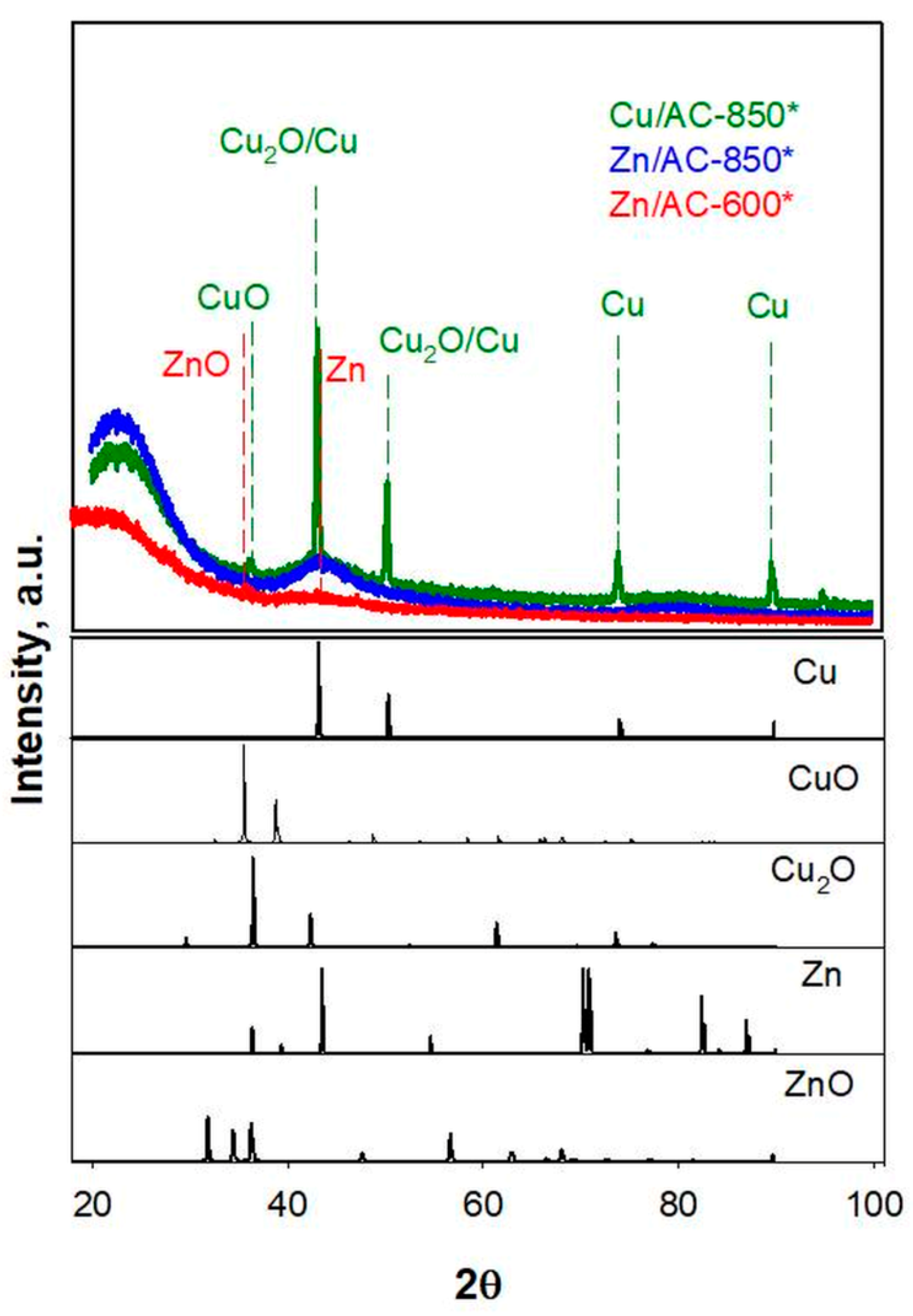
Figure 5 compares XRD patterns of Zn/AC-850*, Zn/AC-600* and Cu/AC-850* sorbents in a 2θ range of 20–100°. All patterns show the broad signals of the amorphous carbon formed upon pyrolysis treatment of PFA. No peaks associable to Zn were observed for Zn/AC-850* sorbents, which is in agreement with Cesano et al. [
10] and with the results of ICP-MS analysis. Small signals assignable to zinc oxide and metallic zinc were detectable in the pattern of Zn/AC-600*, indicating that for lower pyrolysis temperatures some zinc is preserved in the carbon matrix. On the contrary, different peaks with a high intensity, in addition to the carbon background, overlap in the pattern of Cu/Ac-850*, indicating that copper is present in different forms and oxidation states: Cu, CuO, Cu
2O.
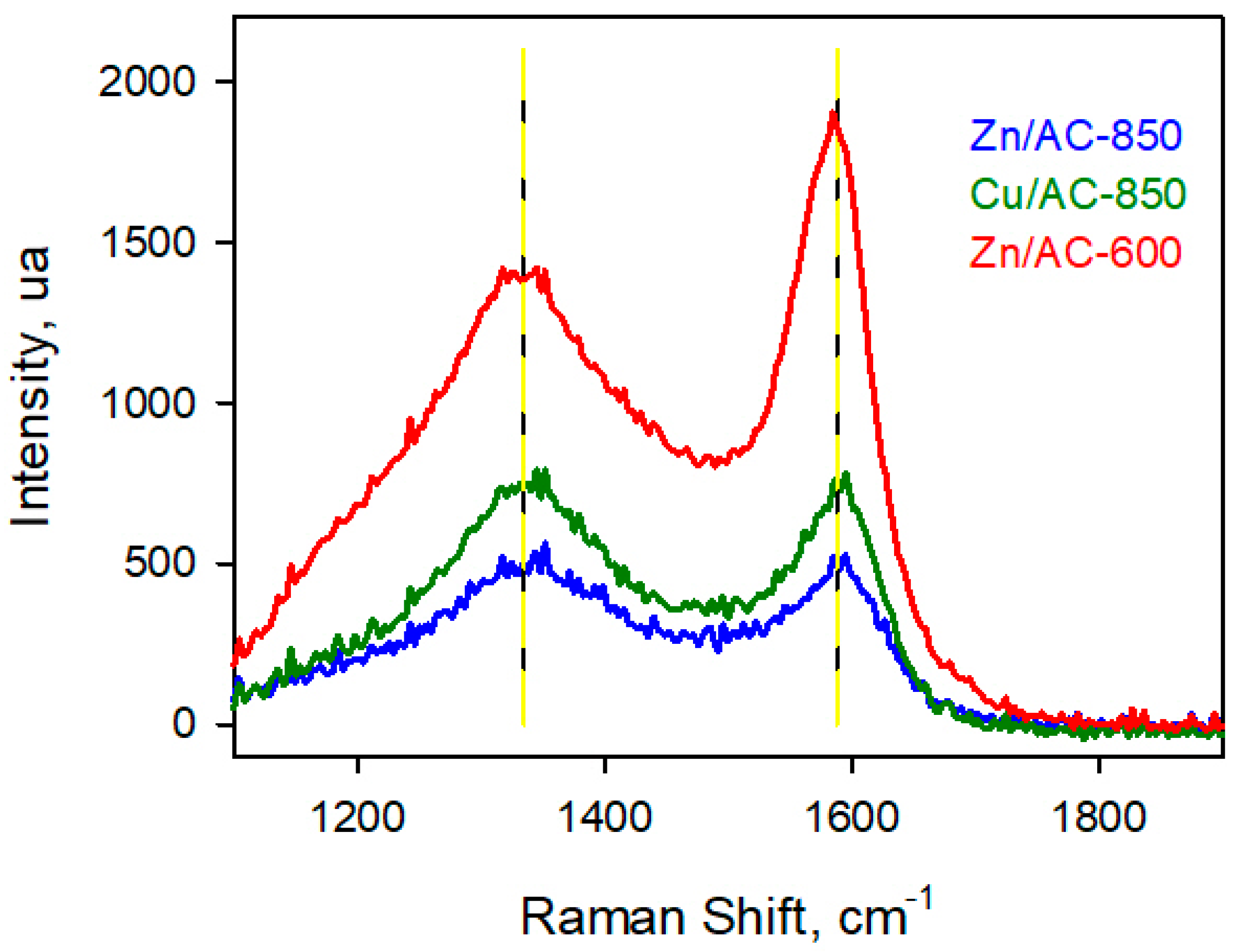
In
Figure 6 the Raman spectra of Zn/AC-850, Zn/Ac-600 and Cu/AC-850 samples are reported in the spectral region 1100–1800 cm
−1, where carbon and its disorder degree can be identified. The Zn-based sample pyrolyzed at 600 °C shows more intense bands and all spectra show the typical D and G bands at 1340 and 1590 cm
−1, assigned to the presence of amorphous and graphitic carbon respectively [
29]. The ratio between the intensities of D and G peaks is considered a common measure of the disorder degree. Ordered carbon materials, such as graphite, show a narrow and intense G band and a wide weak D band, whereas a comparable intensity of D and G band indicates quite a high disorder degree. Values of ratio between the intensity of the two peaks (I(D)/I(G)) are very close to 1 for both samples pyrolyzed at 850 °C, while a value of 0.77 was evaluated for Zn/AC-600. This suggests that a higher pyrolysis temperature partially destroys the graphitic structure that, in turn, is preserved when the temperature is limited to 600 °C.
The porous structure of the AC materials was preliminarily investigated through N
2 physisorption at 77 K. The attempts to explore the 10
−7–10
−5 bar of relative pressure (p/p
0) range for the micropore analysis failed and, for that reason, the investigation was limited to the standard analysis for mesoporous materials. In
Figure 7 the adsorption/desorption isotherms are reported for all the samples pyrolyzed under pure helium (a) and for the corresponding samples pyrolyzed under O
2/He mixture (b). All samples show Type I isotherms, as expected from microporous materials, with a quite well-defined plateau, suggesting a small contribution of the external surface area to the adsorption. A lower temperature of pyrolysis reflects in a much higher porosity for Zn-based AC. Nevertheless, an unusual behaviour was observed for all samples except for Zn/AC-600, i.e., adsorption and desorption branches do not converge to form a closed hysteresis, at least in the range of relative pressure explored, in contrast to what observed by Cesano et al. [
17]. This effect is even more pronounced when oxygen traces are added in the pyrolysis gas.
It is reported that this phenomenon is typically associated with the presence of narrow micropores (width < 1 nm) and/or to pores with “
bottle neck” structure [
30,
31,
32] and that PFA-derived carbons show bottle-like nano- and micropores with narrow openings connected to larger voids [
10].
A similar behaviour was observed for biochar samples and it was explained by the development of a narrow microporosity related to the steam-assisted treatment [
30].
Hysteresis phenomena in pore networks consisting of ink-bottle type pores are quite complex and two basic mechanisms of desorption in these pore networks are known, i.e., pore blocking and cavitation. In the case of the former mechanism, desorption from the pore body may occur only after emptying of its neck. In other words, desorption from the neck triggers evaporation in the blocked pore and the vapor pressure of desorption from the pore body depends on the neck size and network connectivity. Moreover, the presence of narrow micropores represents an obstacle to textural analysis by N
2 adsorption at 77 K independently from the occurrence of bottle neck structures. Indeed, diffusion at 77 K of N
2 molecules in pores having a very small size (i.e., <0.7 nm) is intrinsically limited. For these reasons, the BET-N
2 area does not generally have the physical significance of an effective area if the carbon is ultra-microporous [
22,
30]. According to Morishige et al. [
32], closure of the hysteresis loop takes place at lower relative pressure when the diameter of the pore neck is <5 nm, whereas when the diameter of the neck is >5 nm, the hysteresis loop closes at higher values. As a consequence, since the hysteresis loop did not close at relative p/p
0 pressure as low as 0.1, the presence of pores with a size well below 5 nm can be supposed for our carbons.
With respect to N
2 adsorption at 77 K, CO
2 adsorption at 273 K results in a higher kinetic energy of probe molecules, which can enter into the narrowest pores, thus overcoming both the aforementioned diffusional problems [
7,
31] and limitations related to bottle neck pores [
33]. At 273 K, the CO
2 saturation pressure is quite high, so that the range of p/p
0 is limited to < 0.03 at sub-atmospheric pressures. The initial part of the adsorption isotherm can thus be determined with much greater accuracy than using N
2 at 77 K. In addition, different adsorptives provide complementary information. Finally, it should be noted that the volume of narrow micropores, identified through CO
2 adsorption, was found to be linearly correlated with the hydrogen adsorption capacity of various carbon materials by Jordà-Beneyto et al. [
5] and by Gadiou et al. [
3]. As a consequence, this value can reasonably represent a measure of the H
2 storage properties of this activated carbons.
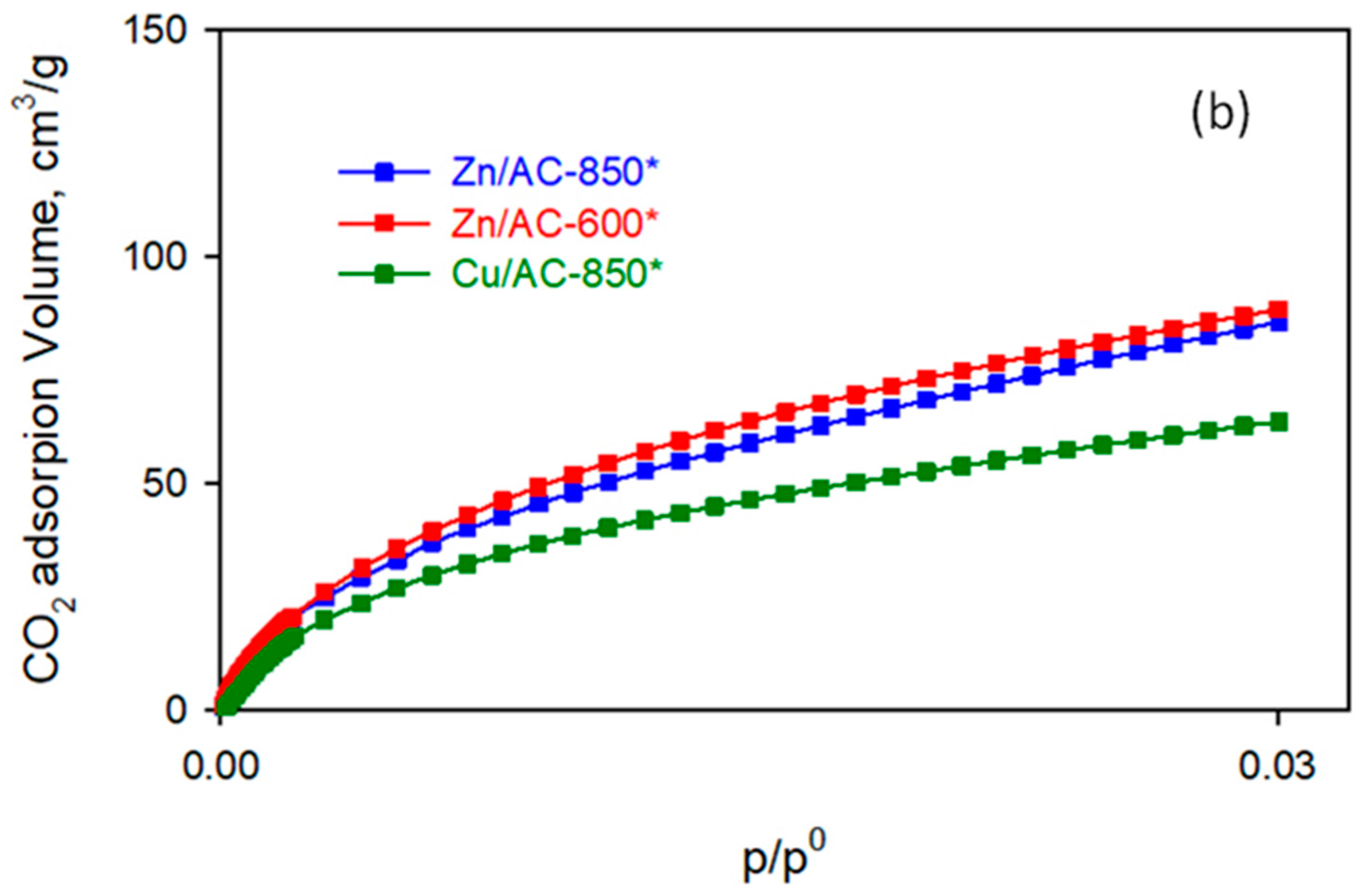
For this reason,
Figure 8 shows CO
2 adsorption isotherms at 273 K on all synthesized AC materials together with the corresponding N
2 adsorption isotherms at 77 K.
Even if limitations in the absolute pressure bearable by the adsorption instrument make it possible to reach only CO
2 relative pressures < 0.03, this kind of data can usually be modelled with the Dubinin–Radushkevich (DR) equation [
34,
35]. Indeed, the DR model allows the extrapolation (as model parameter) of the total micropore volume of the adsorbents, as reported in
Table 3.
The values of the micropore volume, together with those of the characteristic energy of adsorption (another DR model parameter) were used to calculate the effective surface area [
20], pore volume values and mean equivalent radius, in turn reported in
Table 3.
In
Table 3, the surface area values evaluated for the N
2 adsorption branch according to both the BET and DFT models (slit/cylindrical pores model) are also reported together with the total pore volume, estimated at p/p
0 = 0.98 and the cumulative pore volume calculated by the DFT.
Regardless of the pyrolysis conditions reported in
Table 1, all Zn-containing samples show textural parameters that are higher than those reported for similar materials with the same Zn content [
10]. No comparison with literature data can be given for Cu-containing samples due to the novelty in the formulation of these materials.
Values of BET and DFT surface area are quite close for each sample, as well as total pore volume, whereas they are smaller than area and pore volume estimated using the DR model, although only micropore volume was evaluated with this model. Jordà-Beneyto et al. [
5] also observed that pitch-based carbon fibres adsorbed CO
2 at 273 K but did not adsorb N
2 at 77 K, and this suggests that CO
2 adsorption at 273 K makes it possible to also detect pores not accessible through N
2 adsorption at 77 K. Moreover, this supports the choice of the DR model, which is a model suitable for these samples, which are basically microporous with an almost total absence of mesopores.
Nevertheless, although different models were used to estimate the textural parameters of the activated carbons, a unique trend was found for all samples. The results show that Zn-activated PFA derived carbons have a surface area definitively higher than Cu-activated samples, likely due to the quick exothermal polymerization occurring in a few seconds with formation of empty cavities generated by the fast water evaporation. Moreover, surface area increases with both lower pyrolysis temperature and absence of O2 traces in the pyrolysis gas. Therefore, the expected widening of porosity due to the limited burning of carbon did not occur or, if it did occur, it created macropores not evaluated by gas adsorption, likely at the expense of meso and micropores.
Cu-activated carbon shows a much lower surface area and pore volume evaluated according to BET or DFT model, further decreased when oxygen traces are present in the pyrolysis gas, compared to the corresponding Zn-activated samples. Nevertheless, the very large difference observed for both surface area and pore volume using BET or DFT model using N2 physisorption at 77 K between Zn- and Cu-activated carbons is strongly reduced when the surface area and volume are estimated according to the DR model applied to data from CO2 physisorption at 273 K.
This is a reasonable indication of the great contribution of narrow micropores in these samples. Indeed, CO2 is able to identify micropores that are completely undetectable using N2 as adsorbent at 77 K, thus incorrectly identifying Cu-activated carbons as materials with a low porosity.
The latter result is in very good agreement with what was observed by Gargiulo et al. [
30], who found that the pore volume evaluated by N
2 adsorption for bio-chars was lower than that evaluated by CO
2 adsorption, suggesting that very narrow micro-pores were not accessible to N
2 due to diffusional limitation at 77 K. Furthermore, the value of mean average pore size of about 0.75 nm, estimated according to the DR model, confirms that the diameter of pores or of the pore neck is well below 5 nm, as reported by Morishige et al. [
32] and is also in agreement with results of Gargiulo et al. [
30], who found a pore size of around 0.6 nm for their ultra-microporous bio-chars.
In conclusion, the porosimetric results suggest that the two different metal chloride activators lead to carbon materials with rather different textural properties: activated carbons with an almost total dominance of narrow micropores when copper is used as a Lewis acid activator, and activated carbons with meso and micropores when zinc is used as a Lewis acid activator.



 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}