Improving the Activity of Fe/C/N ORR Electrocatalyst Using Double Ammonia Promoted CO2 Laser Pyrolysis

 , ,
, ,
Abstract
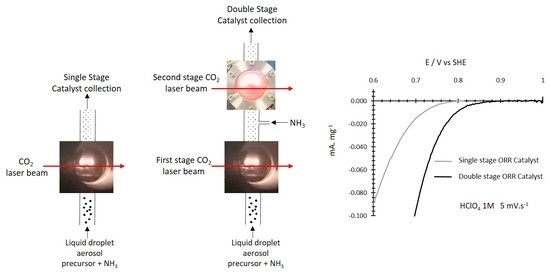
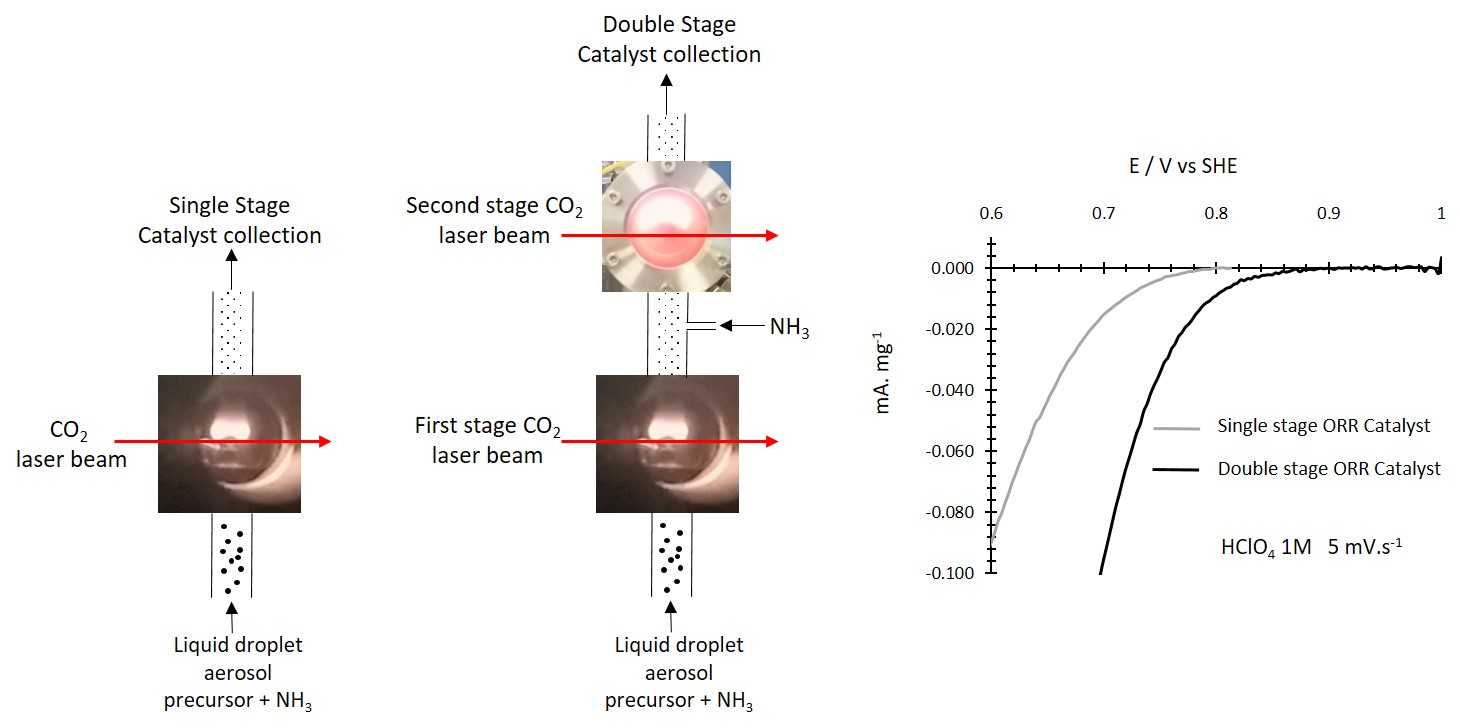
:
1. Introduction
2. Materials and Methods
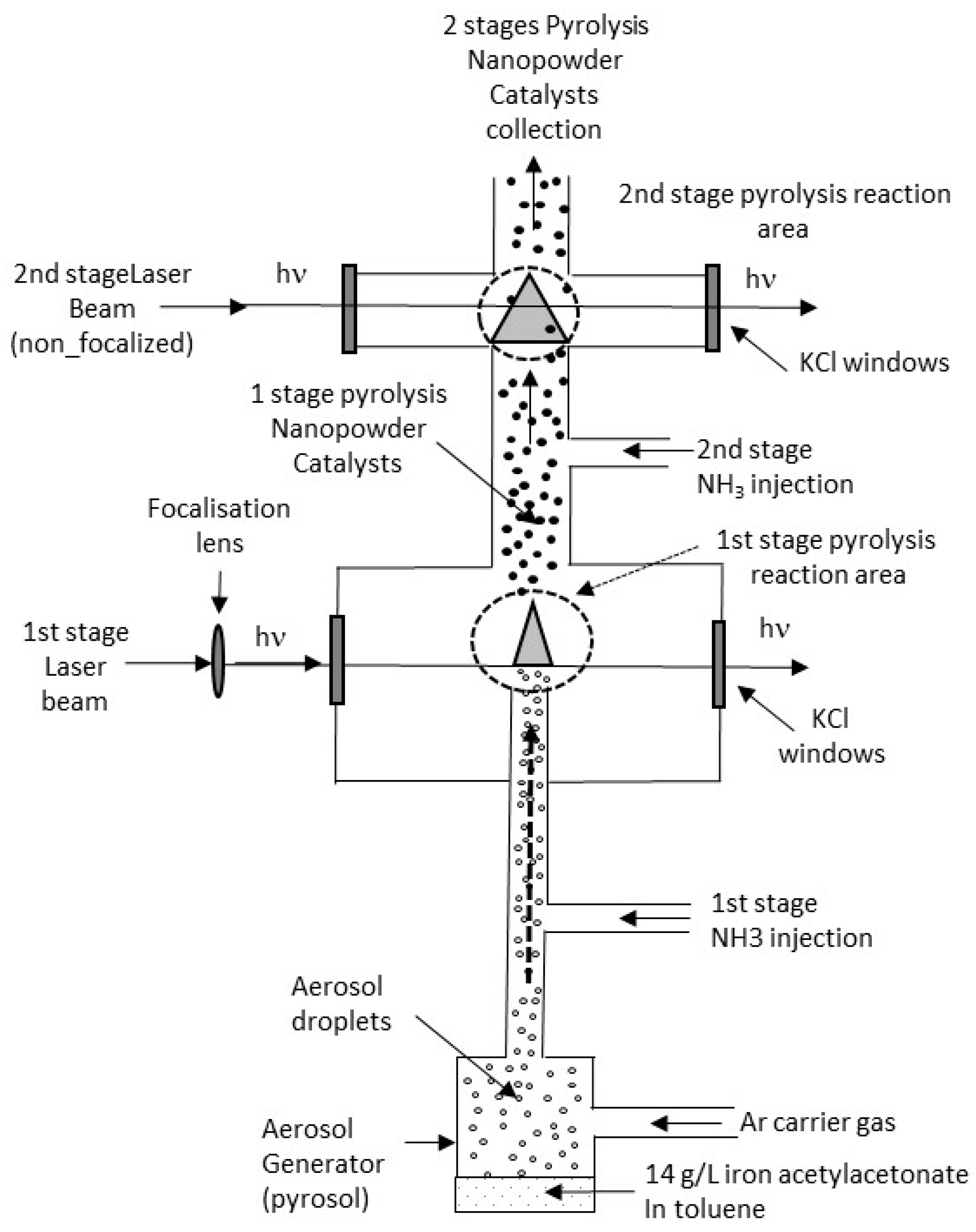
2.1. Synthesis of the Fe/C/N Electrocatalysts
2.2. Characterization of the Fe/C/N Electrocatalysts
2.3. Electrode Preparation and Electrochemical Measurement
3. Results
3.1. Synthesis
3.2. Characterization of the Catalysts
3.2.1. Specific Surface Area, Iron Content, and Morphology
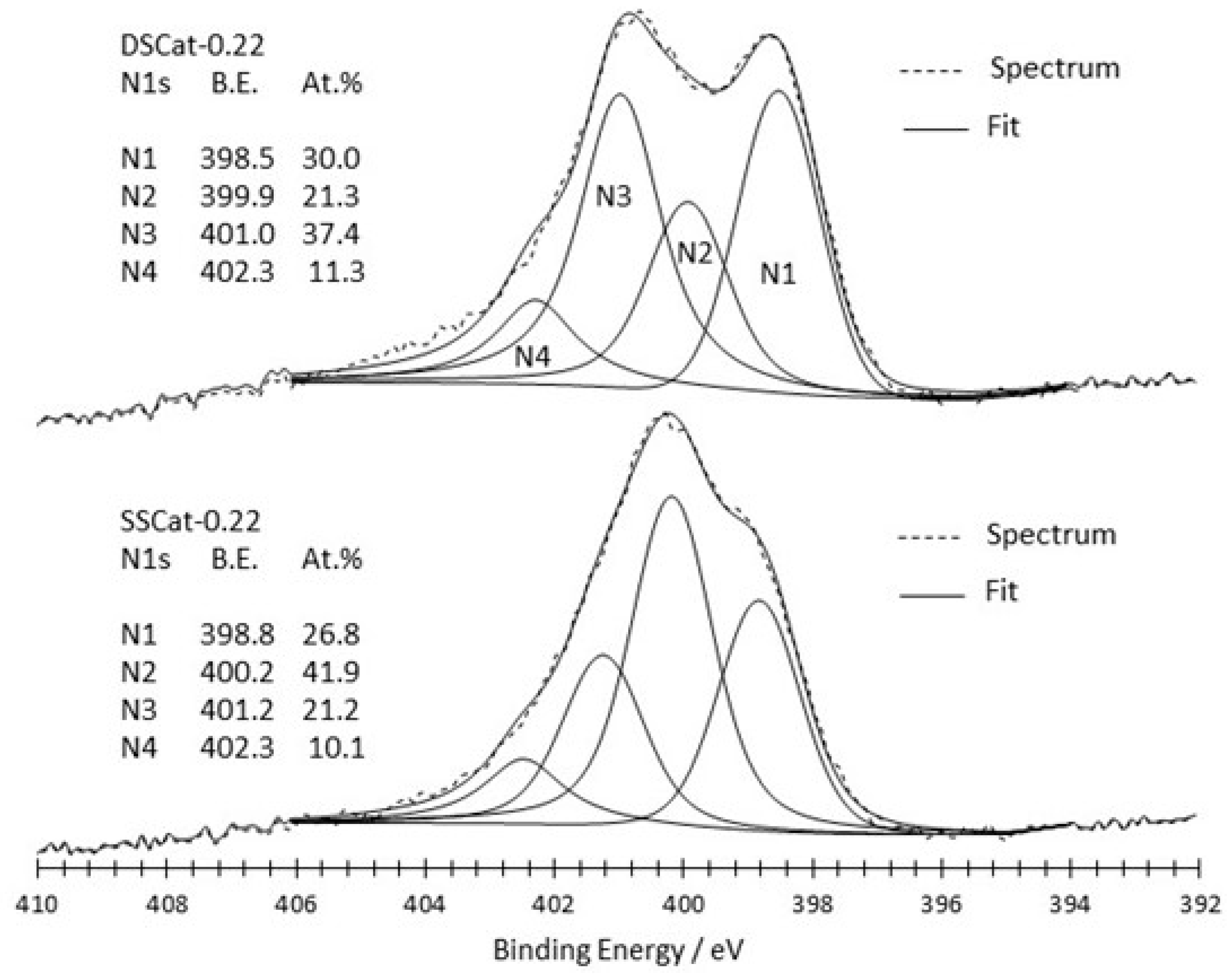
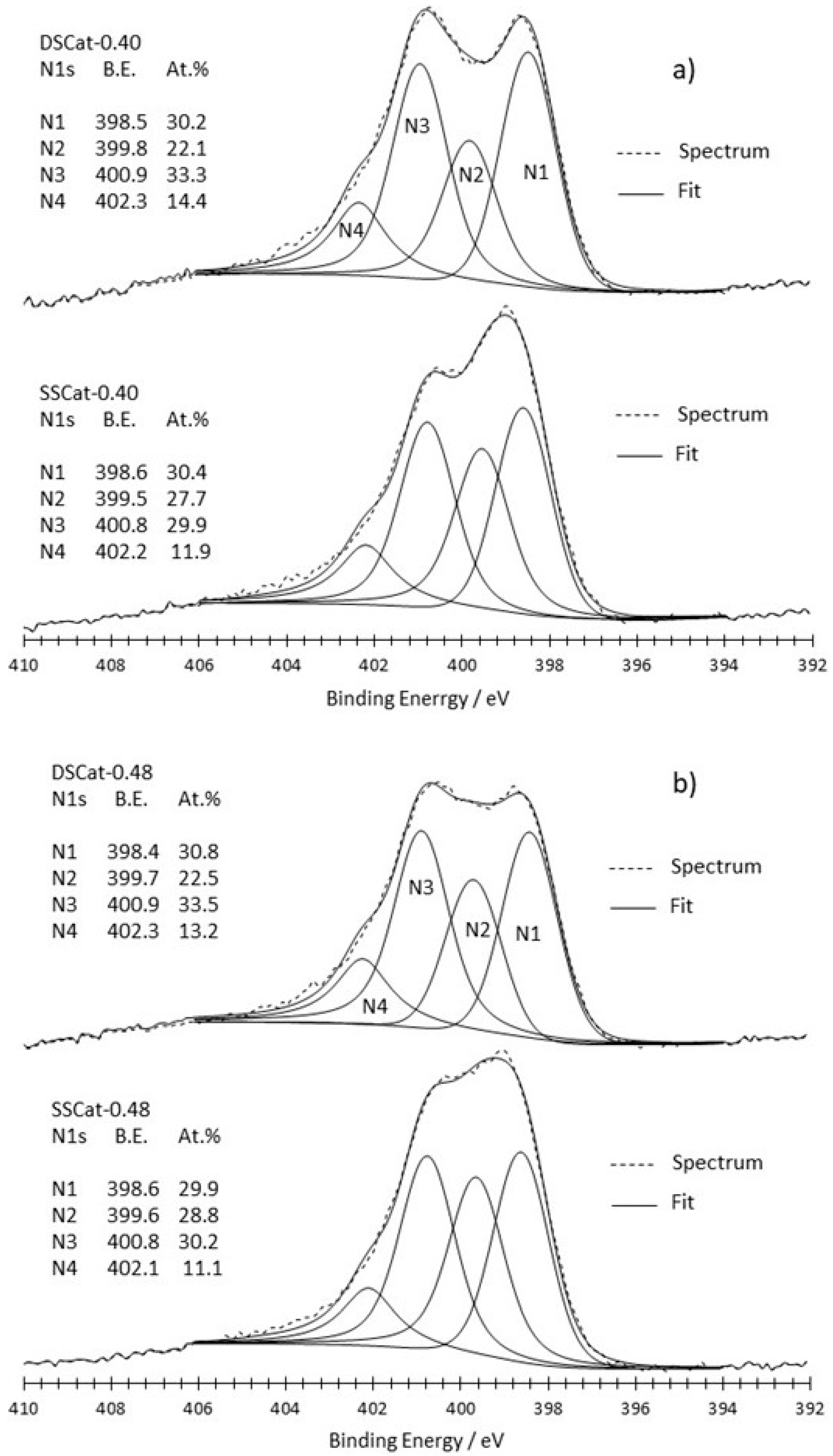
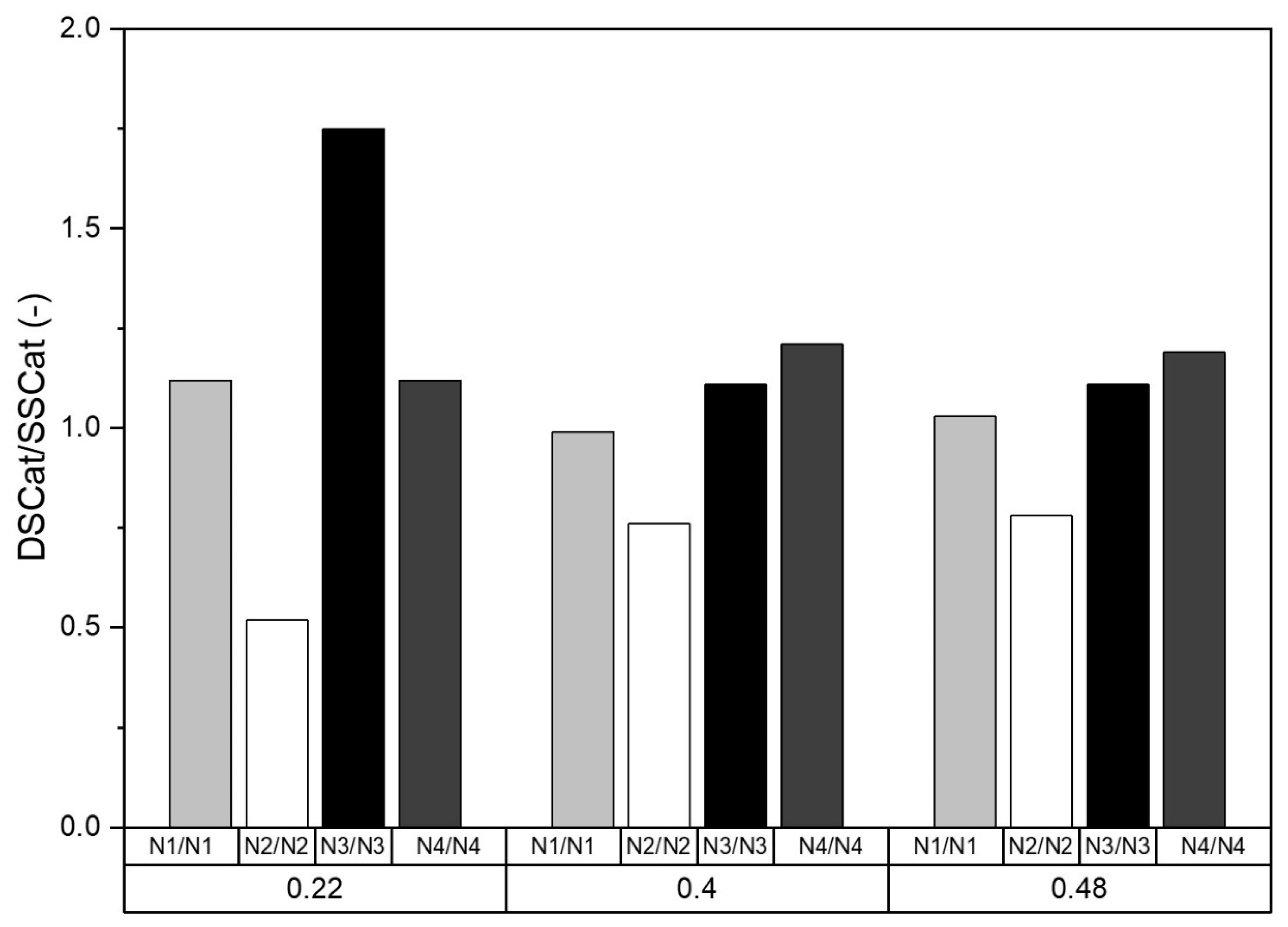
3.2.2. XPS Analysis
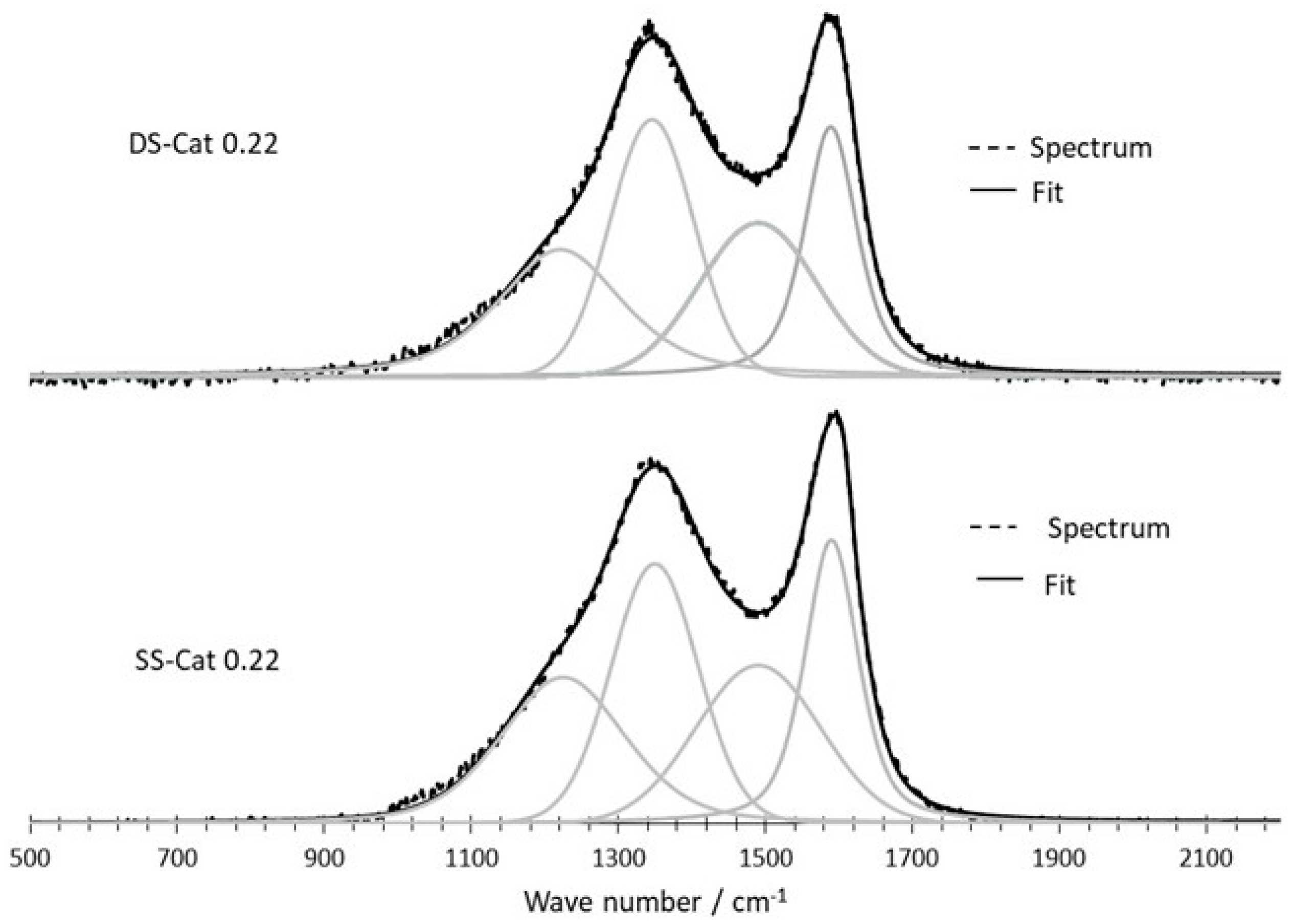
3.2.3. Raman Spectroscopy
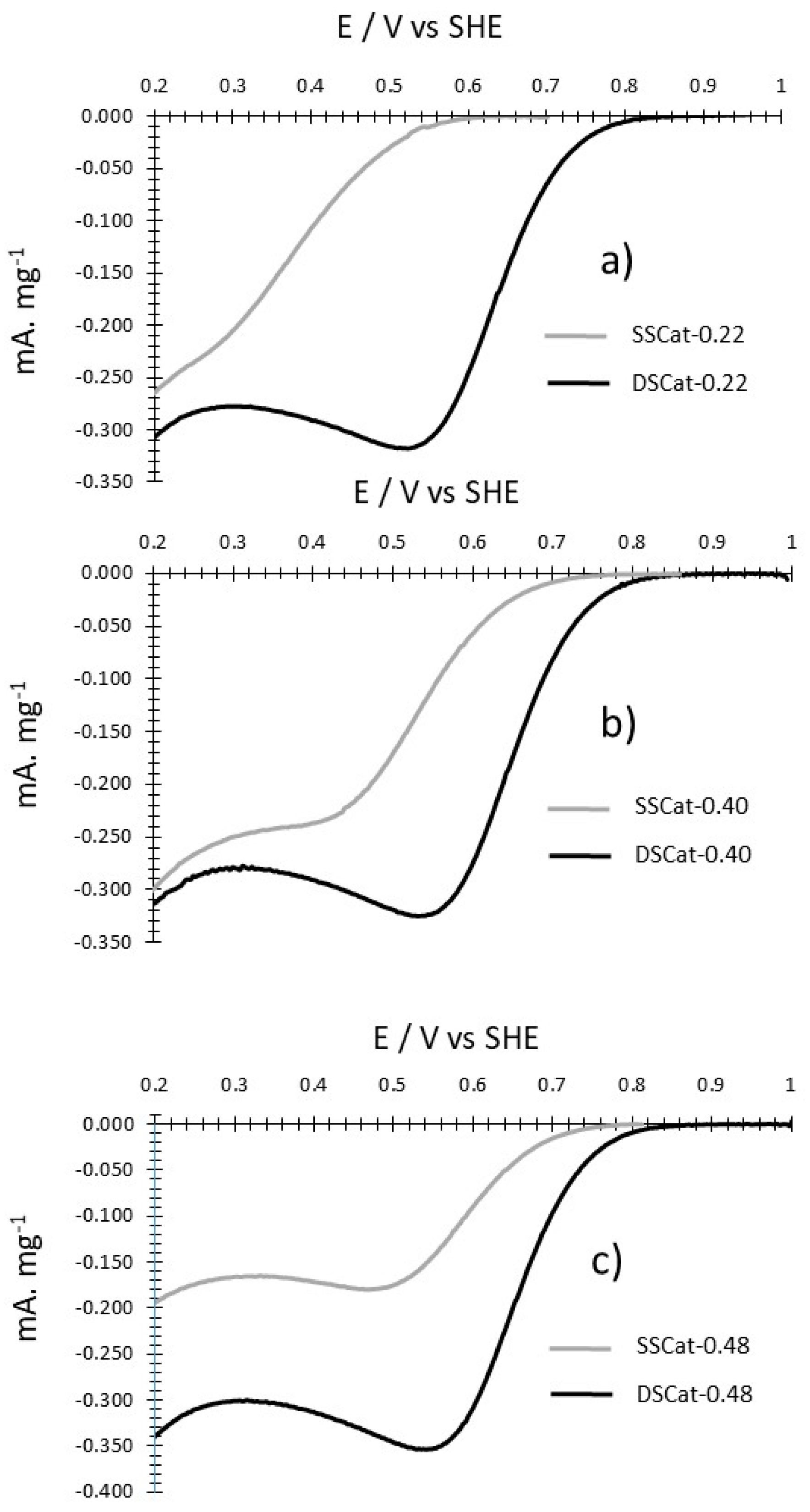
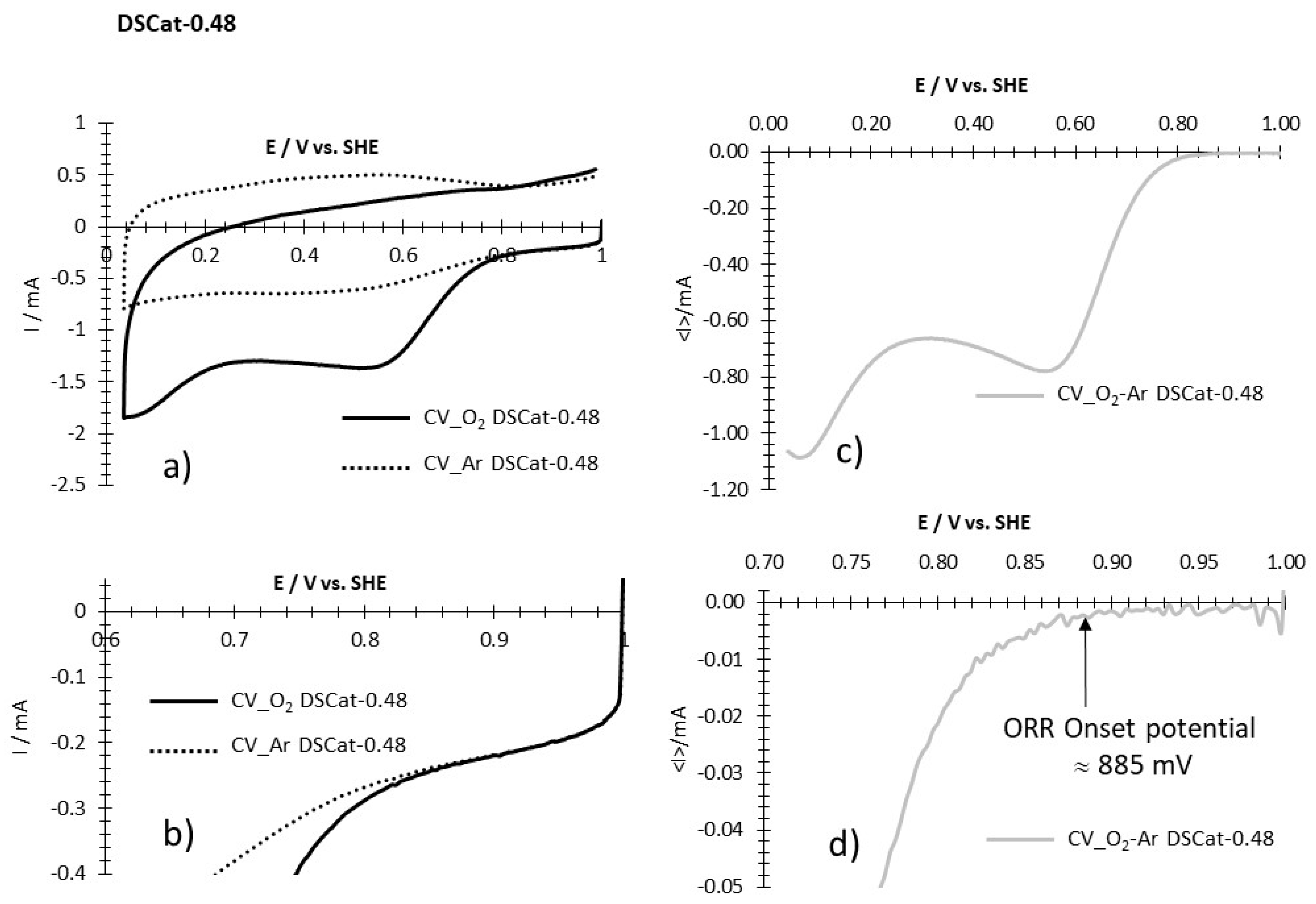
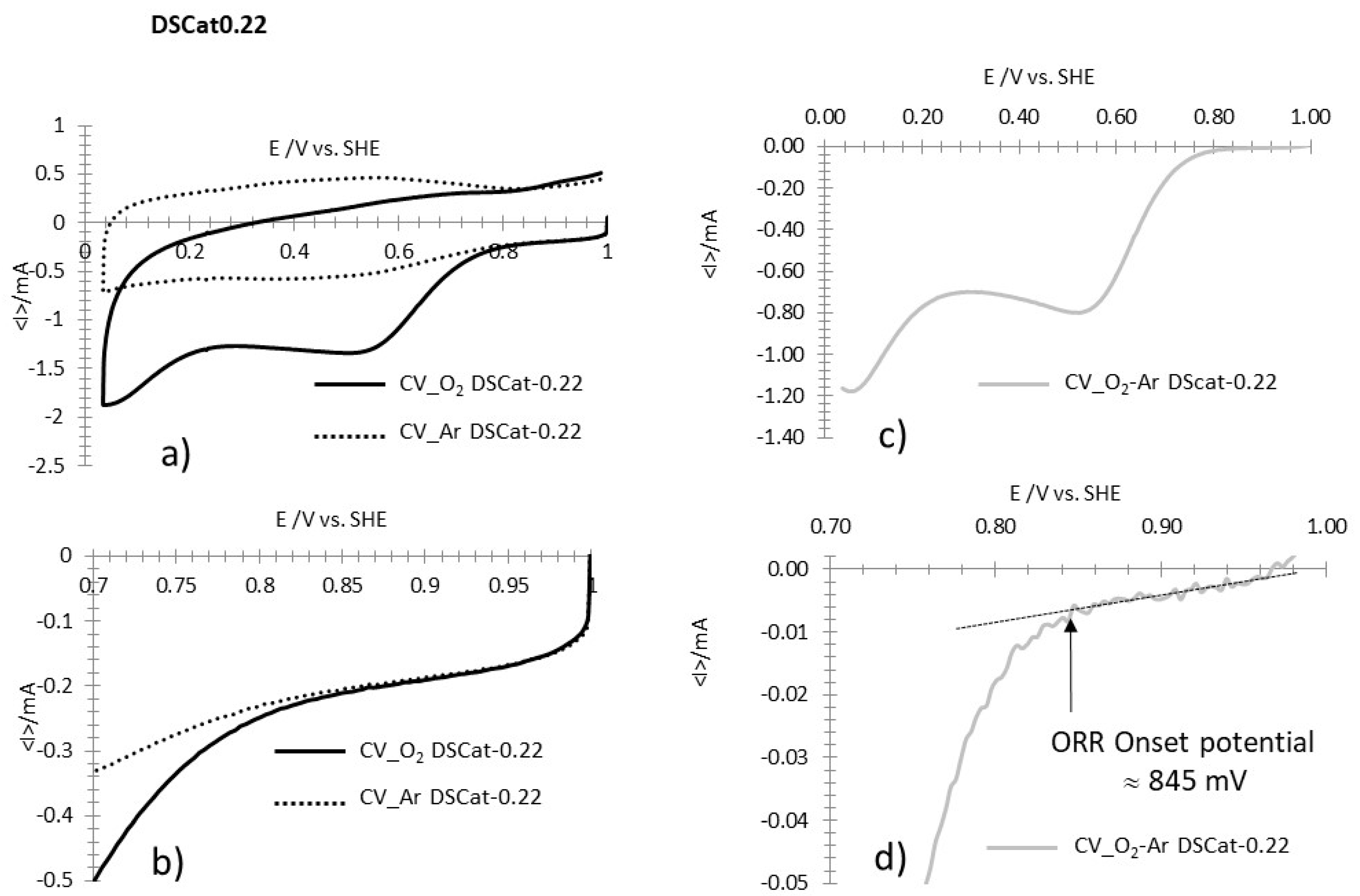
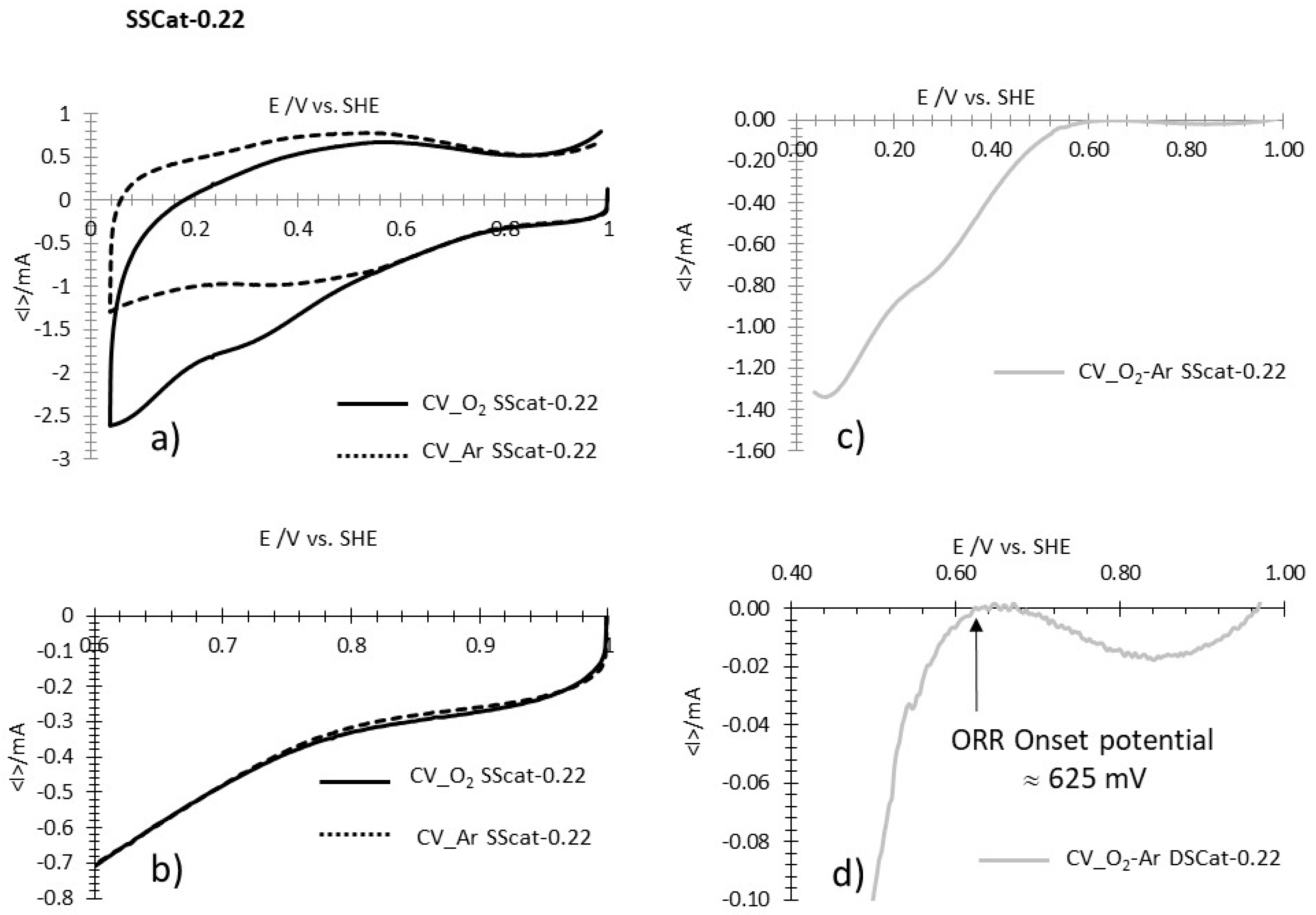
3.2.4. ORR Measurements
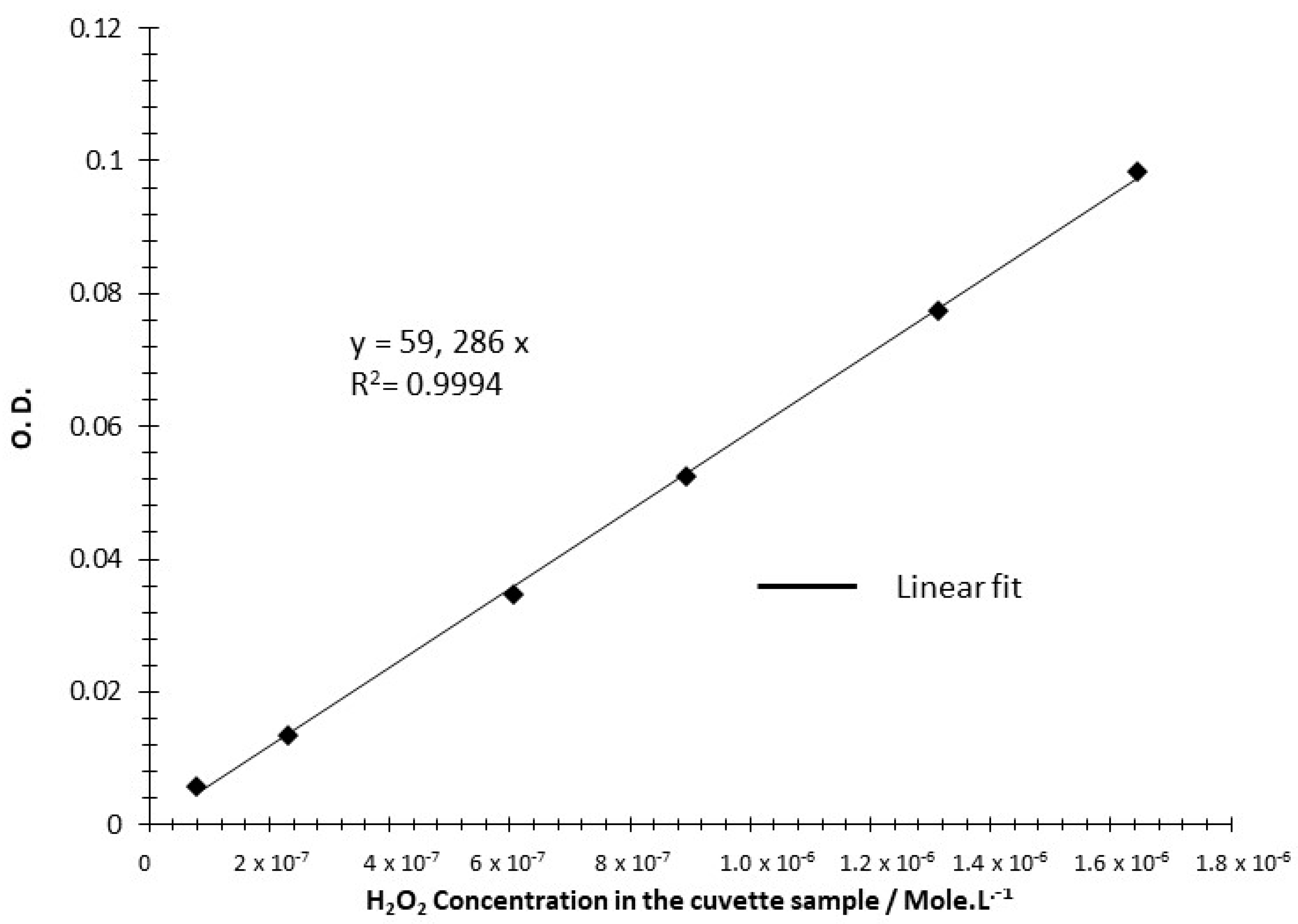
3.2.5. ORR Selectivity Measurement
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A



Appendix B


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SS-Catalysts/Assaying Time | H2O2% | Selectivity | DS-Catalysts/Assaying Time | H2O2% | Selectivity |
|---|---|---|---|---|---|
| SSCat-0.22/10′ | 31.6 | 3.37 | DSCat-0.22/10′ | 24.6 | 3.50 |
| SSCat-0.22/20′ | 29.6 | 3.41 | DSCat-0.22 20′ | 23.0 | 3.54 |
| SSCat-0.22/30′ | 27.3 | 3.45 | DSCat-0.22 20′ | 21.2 | 3.57 |
| SSCat-0.40/10′ | 23.9 | 3.52 | DSCat-0.40/10′ | 14.3 | 3.72 |
| SSCat-0.40/20′ | 22.9 | 3.54 | DSCat-0.40/20′ | 15.1 | 3.70 |
| SSCat-0.40/30′ | 22.2 | 3.56 | DSCat-0.40/30′ | 13.3 | 3.73 |
| SSCat-0.48/10′ | 16.1 | 3.68 | DSCat-0.48/10′ | 12.8 | 3.74 |
| SSCat-0.48/20′ | 16.7 | 3.67 | DSCat-0.48/20′ | 13.7 | 3.72 |
| SSCat-0.48/30′ | 18.0 | 3.64 | DSCat-0.48/30′ | 15.5 | 3.69 |
References
- Osmieri, L. Transition Metal–Nitrogen–Carbon (M–N–C) Catalysts for Oxygen Reduction Reaction. Insights on Synthesis and Performance in Polymer Electrolyte Fuel Cells. Chem. Eng. 2019, 3, 16. [Google Scholar] [CrossRef] [Green Version]
- Ünsala, S.; Yağcı, M.B.; Bozbağa, S.E.; Deljooc, B.; Aindow, M.; Erkeya, C. Supercritical Deposition Coupled with Ammonia Treatment: A New Route to Co Promoted N-doped Carbon Aerogels with High ORR Activity. Energy Technol. 2019, 7, 1900450. [Google Scholar] [CrossRef]
- Shao, M.; Chang, Q.; Dodelet, J.-P.; Chenitz, R. Recent advances in electrocatalysts for oxygen reduction. Chem. Rev. 2016, 116, 3594–3657. [Google Scholar] [CrossRef] [Green Version]
- Banham, D.; Kishimoto, T.; Zhou, Y.; Sato, T.; Bai, K.; Ozaki, J.I.; Imashiro, Y.; Ye, S. Critical advancements in achieving high power and stable non precious metal catalyst-based MEAs for real world proton exchange membrane fuel cell application. Sci. Adv. 2018, 4, eaar7180. [Google Scholar] [CrossRef] [Green Version]
- Proietti, E.; Jaouen, F.; Lefevre, M.; Larouche, N.; Tian, J.; Herranz, J.; Dodelet, J.-P. Iron-based cathode catalyst with enhanced power density in polymer electrolyte membrane fuel cell. Nat. Commun. 2011, 2, 416. [Google Scholar] [CrossRef]
- Goenaga, G.A.; Ma, S.; Yuan, S.; Liu, D.-J. New approaches to Non-PGM electrocatalysts using porous framework materials. ECS Trans. 2010, 33, 579–586. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, X.; Zheng, L.; Shui, J. The Solid-Phase Synthesis of an Fe-N-C Electrocatalyst for High-Power Proton-Exchange Membrane Fuel Cells. Angew. Chem. Int. Ed. 2018, 57, 1204–1208. [Google Scholar] [CrossRef]
- Shen, H.; Gracia-Espino, E.; Ma, J.; Zang, K.; Luo, J.; Wang, L.; Gao, S.; Mamat, X.; Hu, G.; Wagberg, T.; et al. Synergistic Effects between Atomically Dispersed Fe–N–C and C–S–C for the Oxygen Reduction Reaction in Acidic Media. Angew. Chem. Int. Ed. 2017, 56, 13800–13804. [Google Scholar] [CrossRef]
- Longhi, M.; Cova, C.; Pargoletti, E.; Coduri, M.; Santangelo, S.; Patanè, S.; Ditaranto, N.; Cioffi, N.; Facibeni, A.; Scavini, M. Synergistic Effects of Active Sites’ Nature and Hydrophilicity on the Oxygen Reduction Reaction Activity of Pt-Free Catalysts. Nanomaterials 2018, 8, 643. [Google Scholar] [CrossRef] [Green Version]
- Perez, H.; Jorda, V.; Bonville, P.; Vigneron, J.; Frégnaux, M.; Etcheberry, A.; Quinsac, A.; Habert, A.; Leconte, Y. Synthesis and Characterization of Carbon/Nitrogen/Iron Based Nanoparticles by Laser Pyrolysis as Non-Noble Metal Electrocatalysts for Oxygen Reduction. C J. Carbon Res. 2018, 4, 43. [Google Scholar] [CrossRef] [Green Version]
- Perez, H.; Jorda, V.; Vigneron, J.; Frégnaux, M.; Etcheberry, A.; Quinsac, A.; Leconte, Y.; Sublemontier, O. Highly Active, High Specific Surface Area Fe/C/N ORR Electrocatalyst from Liquid Precursors by Combination of CO2 Laser Pyrolysis and Single NH3 Thermal Post-Treatment. C J. Carbon Res. 2019, 5, 26. [Google Scholar] [CrossRef] [Green Version]
- Poozhikunnath, A. Characterization and Optimization of Carbon Based Electrocatalysts and Supports for Fuel Cell Applications. Ph.D. Thesis, University of Connecticut, Storrs, CT, USA, 2019. [Google Scholar]
- Sadezky, A.; Muckenhuber, H.; Grothe, H.; Niessner, R.; Pöschl, U. Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information. Carbon 2005, 43, 1731–1742. [Google Scholar] [CrossRef]
- Cheng, X.; Than, X.-T.; Pinault, M.; Mayne, M.; Reynaud, C.; Vigneron, J.; Etcheberry, A.; Perez, H. Determination of selectivity and specific area related to oxygen reduction reaction as a function of catalyst loading on non-noble metal based electrocatalyst porous electrodes: An example on nitrogen doped carbon nanotube. Electrochim. Acta 2014, 135, 293–300. [Google Scholar] [CrossRef]
- Baret, B.; Aubert, P.-H.; Mayne-L’Hermite, M.; Pinault, M.; Reynaud, C.; Etcheberry, A.; Perez, H. Nanocomposite electrodes based on pre-synthesized organically capped platinum nanoparticles and carbon nanotubes part I: Tuneable low platinum loadings, specific H upd feature and evidence for oxygen reduction. Electrochim. Acta 2009, 54, 5421–5430. [Google Scholar] [CrossRef]
- March, G.; Volatron, F.; Lachaud, F.; Cheng, X.; Baret, B.; Pinault, M.; Etcheberry, A.; Perez, H. Nanocomposite electrodes based on pre-synthesized organically capped platinum nanoparticles and carbon nanotubes. Part II: Determination of diffusion area for oxygen reduction reflects platinum accessibility. Electrochim. Acta 2011, 56, 5151–5157. [Google Scholar] [CrossRef]
- Cheng, X.; Volatron, F.; Pardieu, E.; Borta, A.; Carrot, G.; Reynaud, C.; Mayne, M.; Pinault, M.; Etcheberry, A.; Perez, H. Nanocomposite electrodes based on pre-synthesized organically grafted platinum nanoparticles and carbon nanotubes III. Determination of oxygen reduction reaction selectivity and specific area of porous electrode related to the oxygen reduction reaction ranging from 2 m2 gPt−1 to 310 m2 gPt−1. Electrochim. Acta 2013, 89, 1–12. [Google Scholar] [CrossRef]
- Cheng, X.; Challier, L.; Etcheberry, A.; Noël, V.; Perez, H. The ABTS-HRP system as an alternative method to RRDE for the determination of the selectivity of the oxygen reduction reaction. Int. J. Electrochem. Sci. 2012, 7, 6247–6264. [Google Scholar]
- Artyushkova, K.; Serov, A.; Rojas-Carbonell, S.; Atanassov, P. Chemistry of Multitudinous Active Sites for Oxygen Reduction Reaction in Transition Metal−Nitrogen−Carbon. J. Phys. Chem. C 2015, 119, 25917–25928. [Google Scholar] [CrossRef]
- Primbs, M.; Sun, Y.; Roy, A.; Malko, D.; Mehmood, A.; Sougrati, M.-T.; Blanchard, P.-Y.; Granozzi, G.; Kosmala, T.; Daniel, G.; et al. Establishing reactivity descriptors for platinum group metal (PGM)-free Fe–N–C catalysts for PEM fuel cells. Energy Environ. Sci. 2020. [Google Scholar] [CrossRef]
- Kramm, U.I.; Herranz, J.; Larouche, N.; Arruda, T.M.; Lefevre, M.; Jaouen, F.; Bogdanoff, P.; Fiechter, S.; Abs-Wurmbach, I.; Mukerjee, S.; et al. Structure of the catalytic sites in Fe/N/C-catalysts for O2-reduction in PEM fuel cell. Phys. Chem. Chem. Phys. 2012, 14, 11673–11688. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Zhao, J.P.; Yano, T.; Ooie, T.; Yoneda, M.; Sakakibara, J. Observation of sp3 bonding in tetrahedral amorphous carbon using visible Raman spectroscopy. J. Appl. Phys. 2000, 88, 2305–2308. [Google Scholar] [CrossRef]
- Charon, E.; Rouzaud, J.N.; Aléon, J. Graphitization at low temperatures (600–1200 °C) in the presence of iron implications in planetology. Carbon 2014, 66, 178–190. [Google Scholar] [CrossRef]
- Luo, E.; Wang, C.; Li, Y.; Wang, X.; Gong, L.; Zhao, T.; Jin, Z.; Ge, J.; Liu, C.; Xing, W. Accelerated oxygen reduction on Fe/N/C catalysts derived from precisely-designed ZIF precursors. Nano Res. 2020, 13, 2420–2426. [Google Scholar] [CrossRef]
- Lo Vecchio, C.; Arico, A.S.; Monforte, G.; Baglio, V. EDTA-derived CoeNeC and FeeNeC electro-catalysts for the oxygen reduction reaction in acid environment. Renew. Energy 2018, 120, 342–349. [Google Scholar] [CrossRef]
- Jimenez-Mateos, J.M.; Fiero, J.L.G. X-ray Photoelectron Spectroscopic Study of Petroleum Fuel Cokes. Surf. Interface Anal. 1996, 24, 223–226. [Google Scholar] [CrossRef]
- Casanovas, J.; Ricart, J.M.; Rubio, J.; Illas, F.; Jimenez-Mateos, J.M. Origin of the Large N 1s Binding Energy in X-ray Photoelectron Spectra of Calcined Carbonaceous Materials. J. Am. Chem. Soc. 1996, 118, 8071–8076. [Google Scholar] [CrossRef]
- Ding, W.; Wei, Z.; Chen, S.; Qi, X.; Yang, T.; Hu, J.; Wang, D.; Wan, L.-J.; Fatima Alvi, S.F.; Li, L. Nitrogen Space-Confinement-Induced Synthesis of Pyridinic- and Pyrrolic Nitrogen-Doped Graphene for the Catalysis of Oxygen Reduction. Angew. Chem. Int. Ed. 2013, 52, 11755–11759. [Google Scholar] [CrossRef]
- Pels, J.R.; Kapteijn, F.; Moulijn, J.A.; Zhu, Q.; Thomas, K.M. Evolution of nitrogen functionalities in carbonaceous materials during pyrolysis. Carbon 1995, 33, 1641–1653. [Google Scholar] [CrossRef]
- Arrigo, R.; Hävecker, M.; Schlög, R.; Sheng Su, D. Dynamic surface rearrangement and thermal stability of nitrogen functional groups on carbon nanotubes. Chem. Commun. 2008, 40, 4891–4893. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |








| Catalyst | Catalyst Production Rate As-Formed g·h−1 | Chemical Yield As-Formed Material wt% | Catalyst Production Rate after Acetone Washing g·h−1 | Chemical Yield after Acetone Washing wt% |
|---|---|---|---|---|
| SSCat-0.22 | 8.8 | 28.0 | 6.9 | 21.9 |
| SSCat-0.40 | 7.5 | 23.9 | 6.0 | 19.1 |
| SSCat-0.48 | 6.0 | 19.0 | 4.4 | 14.1 |
| DSCat-0.22 | 6.1 | 19.5 | 5.8 | 18.3 |
| DSCat-0.40 | 5.1 | 16.1 | 4.8 | 15.2 |
| DSCat-0.48 | 3.5 | 10.9 | 3.1 | 9.8 |
| Catalyst | Iron Content wt% | Specific Surface Area m2·g−1 |
|---|---|---|
| SSCat-0.22 | 0.65 | 124 |
| SSCat-0.40 | 0.83 | 135 |
| SSCat-0.48 | 1.03 | 159 |
| DSCat-0.22 | 0.41 | 115 |
| DSCat-0.40 | 0.59 | 144 |
| DSCat-0.48 | 1.00 | 172 |
| Catalyst | C1s at.% | O1s at.% | N1s at.% | N1s at.%/C1s at.% |
|---|---|---|---|---|
| SSCat-0.22 | 93.6 | 4.4 | 2.0 | 0.021 |
| SSCat-0.40 | 92.2 | 5.6 | 2.2 | 0.024 |
| SSCat-0.48 | 92.4 | 4.4 | 3.2 | 0.034 |
| DSCat-0.22 | 94.8 | 3.1 | 2.1 | 0.022 |
| DSCat-0.40 | 94.5 | 2.7 | 2.8 | 0.028 |
| DSCat-0.48 | 93.7 | 2.9 | 3.4 | 0.030 |
| R = 0.22 | R = 0.40 | R = 0.48 | |
|---|---|---|---|
| ID/IG SS-Cat | 0.90 ± 0.06 | 0.94 ± 0.06 | 0.88 ± 0.04 |
| ID/IG DSCat | 1.03 ± 0.05 | 0.98 ± 0.05 | 0.95 ± 0.05 |
| Catalyst | SSCat-0.22 | SSCat-0.40 | SSCat0.48 | DSCat-0.22 | DSCat-0.40 | DSCat-0.48 |
|---|---|---|---|---|---|---|
| H2O2% | 29.5 | 23.0 | 16.9 | 22.9 | 14.2 | 14.0 |
| Selectivity | 3.41 | 3.54 | 3.66 | 3.54 | 3.72 | 3.72 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez, H.; Frégnaux, M.; Charon, E.; Etcheberry, A.; Sublemontier, O. Improving the Activity of Fe/C/N ORR Electrocatalyst Using Double Ammonia Promoted CO2 Laser Pyrolysis. C 2020, 6, 63. https://doi.org/10.3390/c6040063
Perez H, Frégnaux M, Charon E, Etcheberry A, Sublemontier O. Improving the Activity of Fe/C/N ORR Electrocatalyst Using Double Ammonia Promoted CO2 Laser Pyrolysis. C. 2020; 6(4):63. https://doi.org/10.3390/c6040063
Chicago/Turabian StylePerez, Henri, Mathieu Frégnaux, Emeline Charon, Arnaud Etcheberry, and Olivier Sublemontier. 2020. "Improving the Activity of Fe/C/N ORR Electrocatalyst Using Double Ammonia Promoted CO2 Laser Pyrolysis" C 6, no. 4: 63. https://doi.org/10.3390/c6040063
APA StylePerez, H., Frégnaux, M., Charon, E., Etcheberry, A., & Sublemontier, O. (2020). Improving the Activity of Fe/C/N ORR Electrocatalyst Using Double Ammonia Promoted CO2 Laser Pyrolysis. C, 6(4), 63. https://doi.org/10.3390/c6040063