



Molecules 2014, 19(5), 6450-6473; https://doi.org/10.3390/molecules19056450 - 20 May 2014
Cited by 43 | Viewed by 9783
Abstract
►
Show Figures
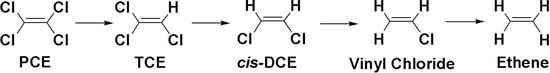
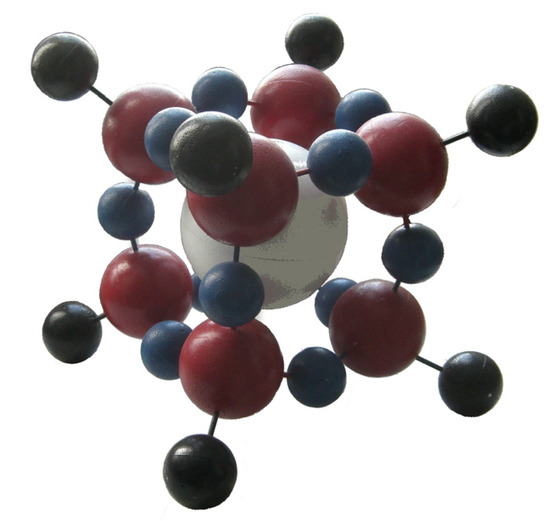
Chlorinated ethenes are prevalent groundwater contaminants. To better constrain (bio)chemical reaction mechanisms of reductive dechlorination, the position-specificity of reductive trichloroethene (TCE) dehalogenation was investigated. Selective biotransformation reactions (i) of tetrachloroethene (PCE) to TCE in cultures of Desulfitobacterium sp. strain Viet1; and (ii) of
[...] Read more.
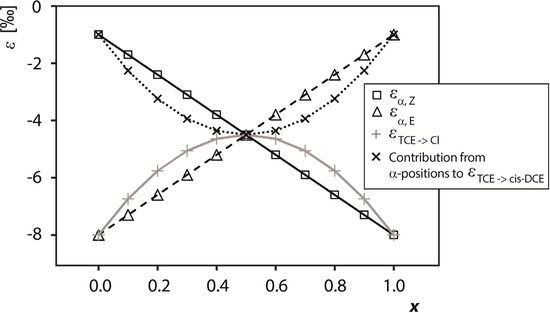
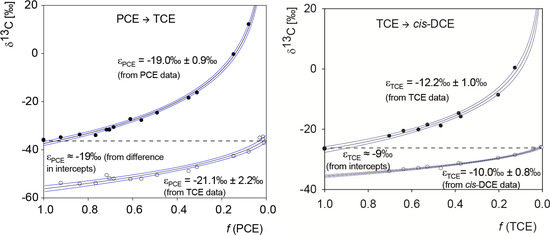
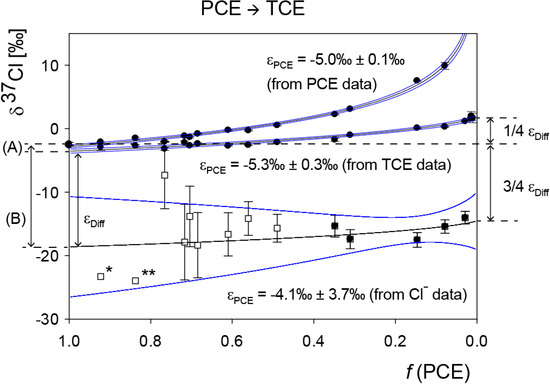
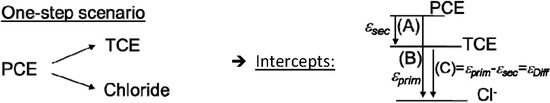
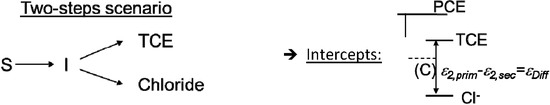
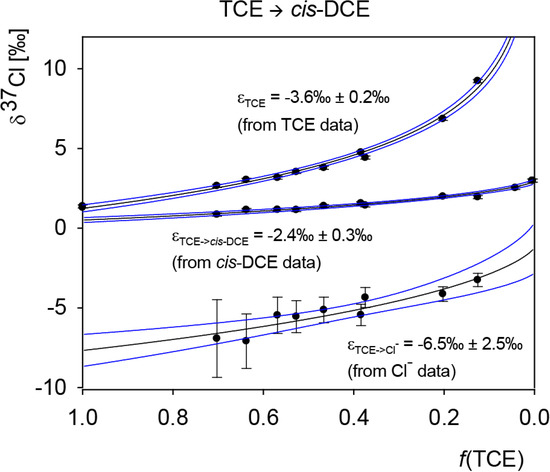
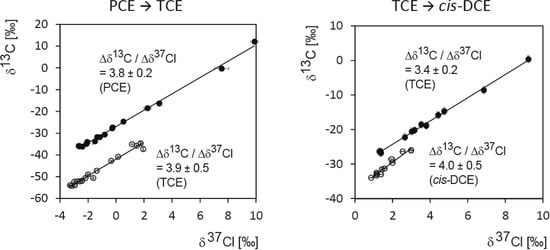
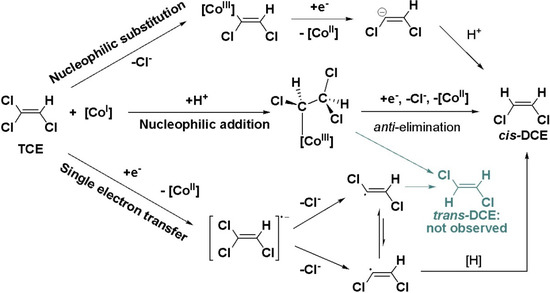
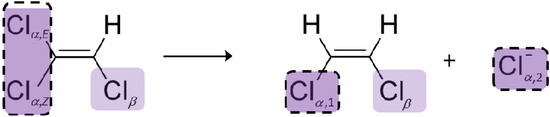
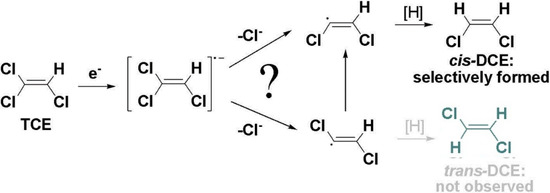
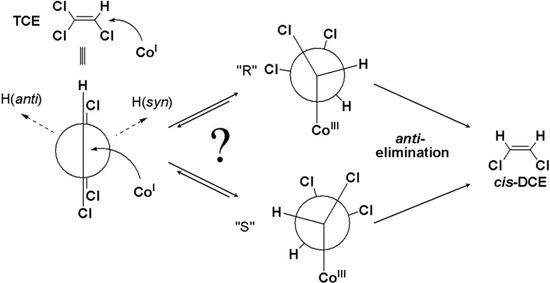
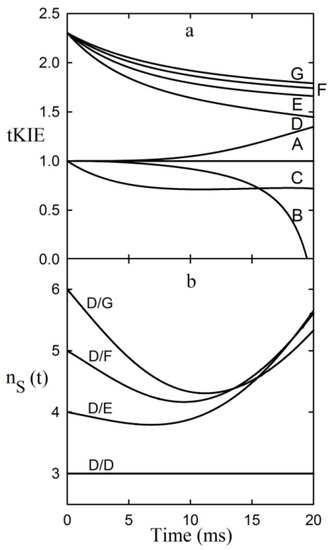
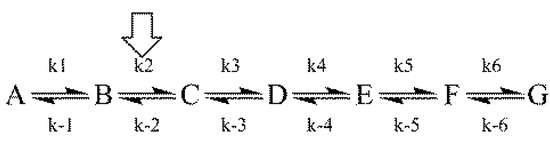
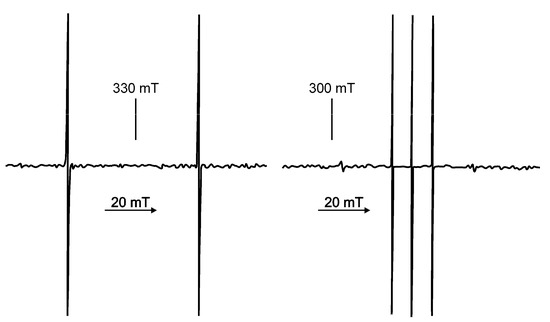
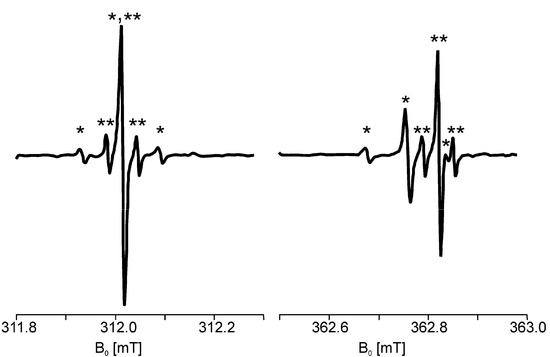
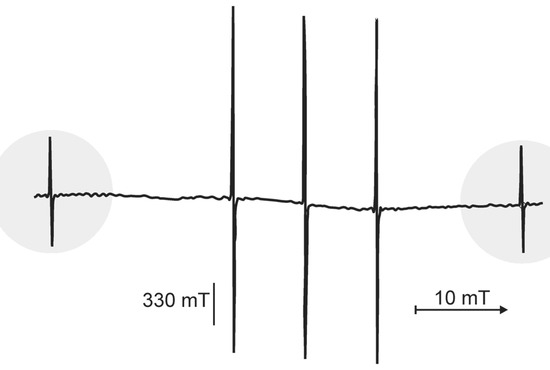
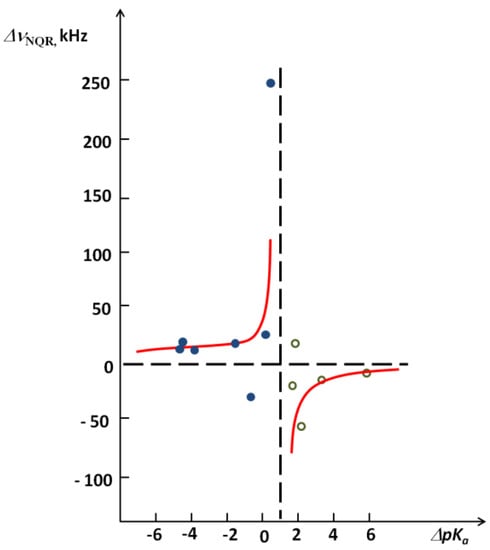
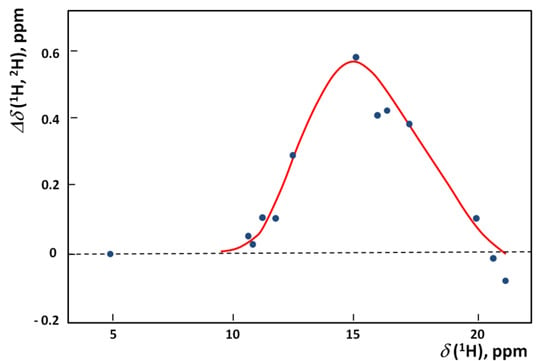
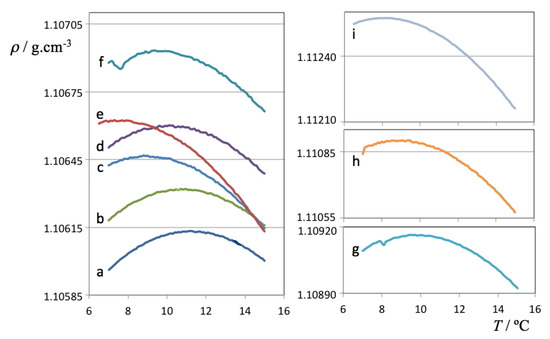
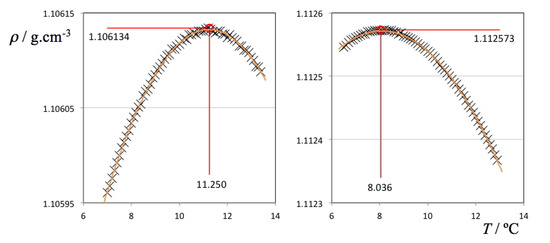
Chlorinated ethenes are prevalent groundwater contaminants. To better constrain (bio)chemical reaction mechanisms of reductive dechlorination, the position-specificity of reductive trichloroethene (TCE) dehalogenation was investigated. Selective biotransformation reactions (i) of tetrachloroethene (PCE) to TCE in cultures of Desulfitobacterium sp. strain Viet1; and (ii) of TCE to cis-1,2-dichloroethene (cis-DCE) in cultures of Geobacter lovleyi strain SZ were investigated. Compound-average carbon isotope effects were −19.0‰ ± 0.9‰ (PCE) and −12.2‰ ± 1.0‰ (TCE) (95% confidence intervals). Using instrumental advances in chlorine isotope analysis by continuous flow isotope ratio mass spectrometry, compound-average chorine isotope effects were measured for PCE (−5.0‰ ± 0.1‰) and TCE (−3.6‰ ± 0.2‰). In addition, position-specific kinetic chlorine isotope effects were determined from fits of reactant and product isotope ratios. In PCE biodegradation, primary chlorine isotope effects were substantially larger (by −16.3‰ ± 1.4‰ (standard error)) than secondary. In TCE biodegradation, in contrast, the product cis-DCE reflected an average isotope effect of −2.4‰ ± 0.3‰ and the product chloride an isotope effect of −6.5‰ ± 2.5‰, in the original positions of TCE from which the products were formed (95% confidence intervals). A greater difference would be expected for a position-specific reaction (chloride would exclusively reflect a primary isotope effect). These results therefore suggest that both vicinal chlorine substituents of TCE were reactive (intramolecular competition). This finding puts new constraints on mechanistic scenarios and favours either nucleophilic addition by Co(I) or single electron transfer as reductive dehalogenation mechanisms.
Full article
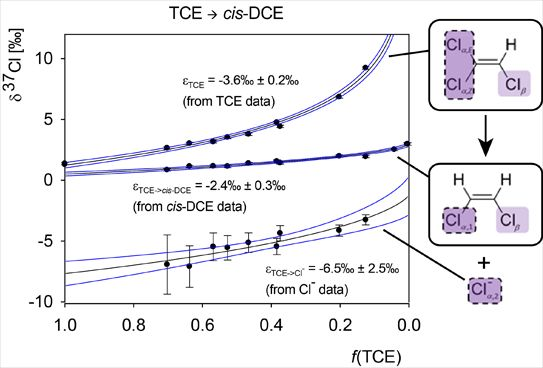
Graphical abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}