


Thalass. Rep. 2025, 15(1), 1; https://doi.org/10.3390/thalassrep15010001 - 31 Jan 2025
Abstract
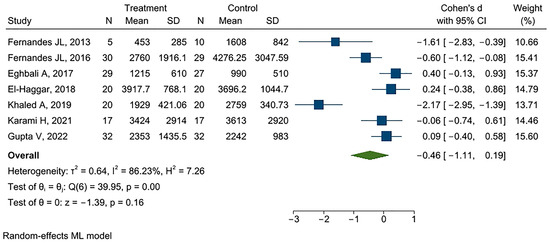
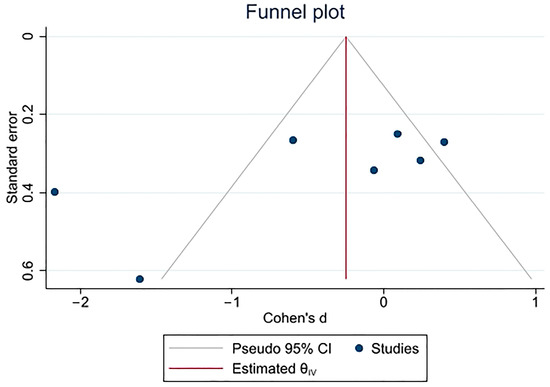
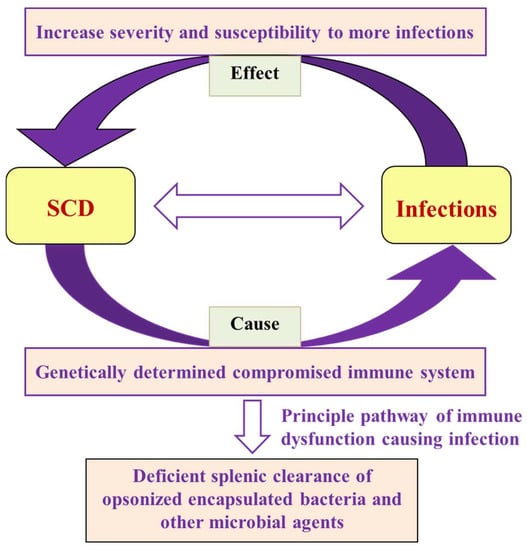
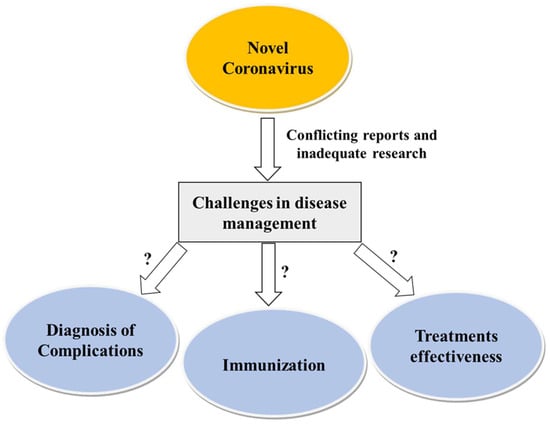
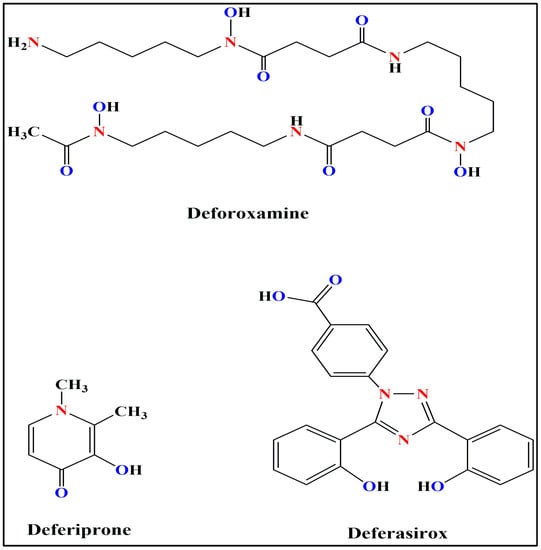
It is important to recognize the significance of acute painful transfusion reactions (APTRs) as a complication of blood transfusions. These adverse reactions are characterized by the onset of acute pain after the administration of blood components. Despite their potential to cause significant discomfort
[...] Read more.
It is important to recognize the significance of acute painful transfusion reactions (APTRs) as a complication of blood transfusions. These adverse reactions are characterized by the onset of acute pain after the administration of blood components. Despite their potential to cause significant discomfort and distress to patients, these reactions are frequently disregarded and under-reported in clinical practice. This review aims to provide a comprehensive analysis of the clinical features, pathophysiology, diagnosis, incidence, and management of APTRs, emphasizing the necessity for heightened awareness and further research in this field.
Full article
(This article belongs to the Section Innovative Treatment of Thalassemia)
►
Show Figures

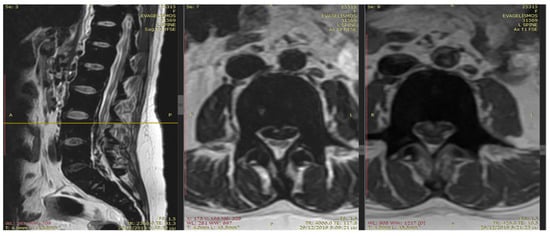
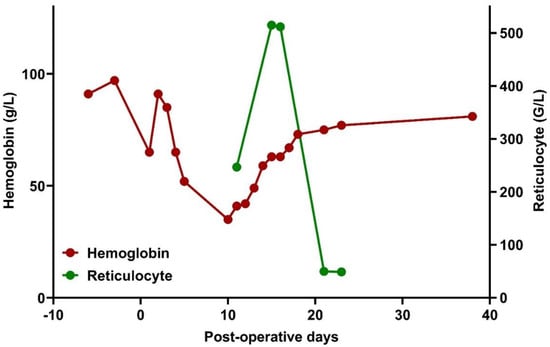
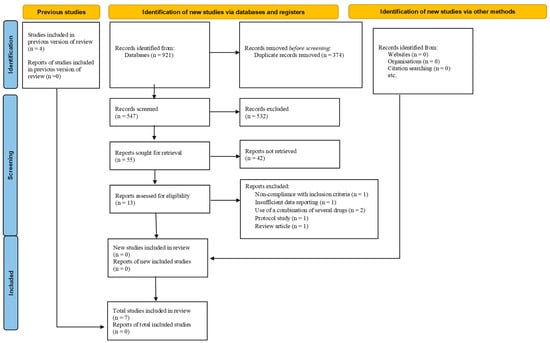
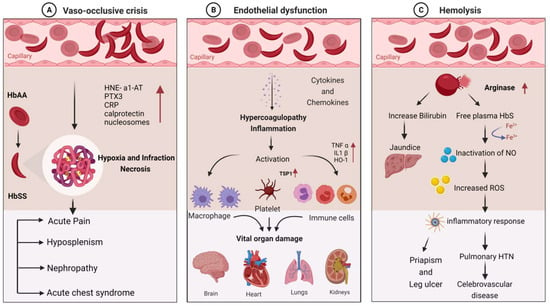
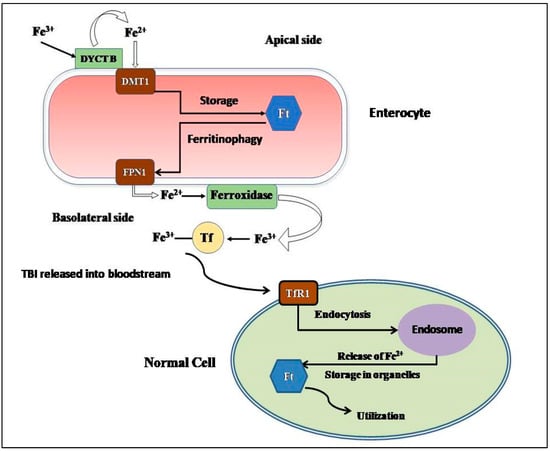
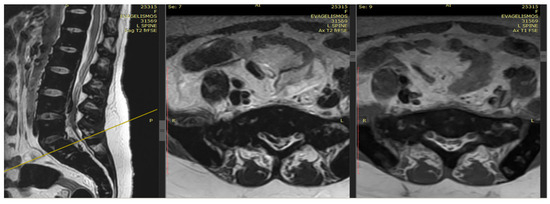
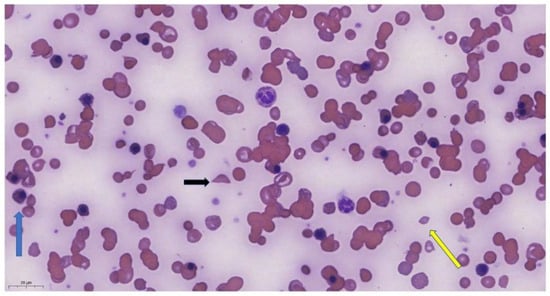
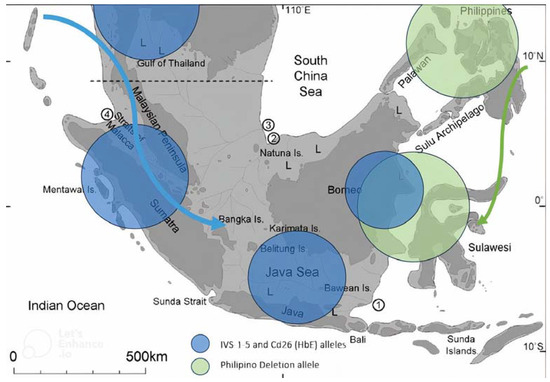
Figure 1











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}