New Paracyclophanylthiazoles with Anti-Leukemia Activity: Design, Synthesis, Molecular Docking, and Mechanistic Studies

 ,
,  , and
, and
Abstract
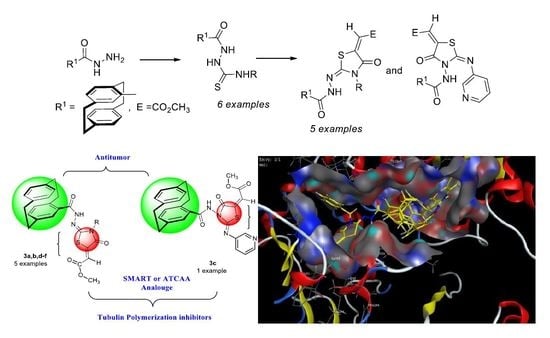
:
Highlights
- -
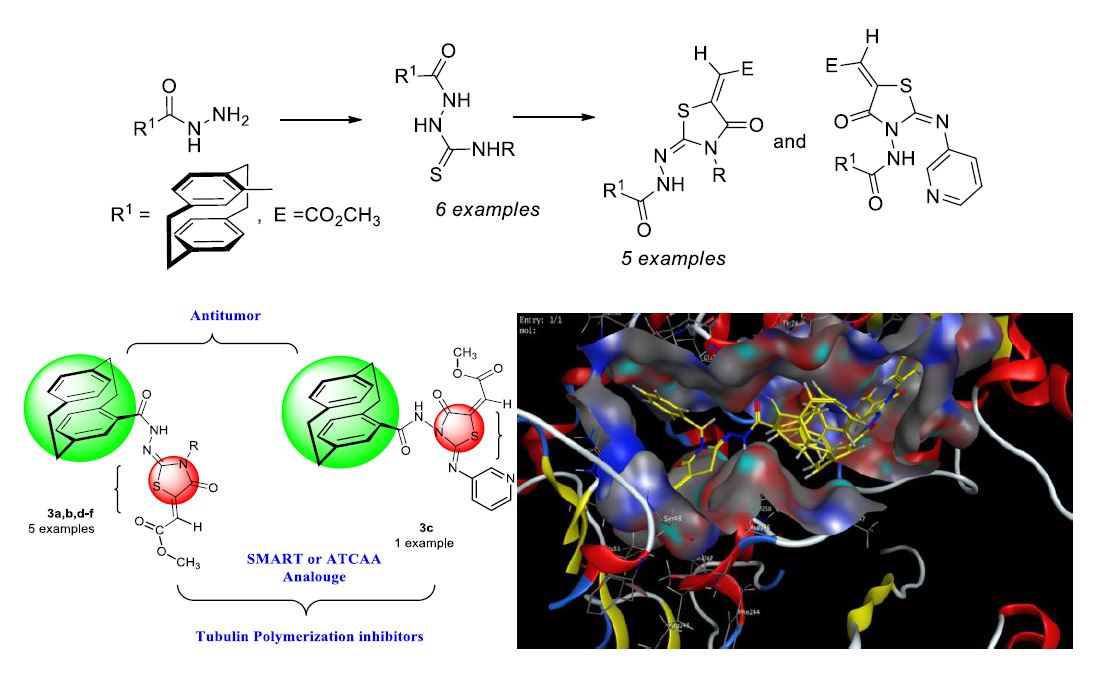
- Synthesis of methyl 2-(2-(4′-[2.2]paracyclophanyl)-hydrazine-ylidene)-4-oxothiazolidin-5-ylidene)acetates was here reported.
- -
- Cytotoxic activity of the synthesized compounds toward the NCI-60 panel of cancer cell lines was determined, and the cellular mechanism of the most potent inhibitors was further investigated in leukemia cell lines.
- -
- Compound 3a was found as the most cytotoxic one, and it was selected for further five-dose analysis according to NCI protocol.
- -
- Compound 3a, in comparison to other derivatives, exhibited high specificity against leukemia RPMI-8226 and SR cell lines, and it also showed a remarkable tubulin inhibitory activity in relation to colchicine with IC50 = 4.97 µM/mL.
- -
- Docking studies with β-tubulin revealed that most of the tested compounds showed good binding at the colchicine binding site of the enzyme, especially for compound 3a, which made several interactions better than that of the reference colchicine.
1. Introduction
2. Results and Discussion
2.1. Chemistry Section
Preparation of 2-(4′-[2.2]Paracyclophanyl-4H-hydrazinecarbothioamides 2a–f
2.2. Biological Investigation
2.2.1. Anti-Proliferative Investigation against 60 Cancer Cell Lines at the National Cancer Institute (NCI), USA.
Structure Activity Relationship (SAR)
2.2.2. In Vitro Five-Dose Full NCI 60 Cell Panel Assay
2.2.3. Evaluation of In Vitro Antiproliferative Activities against Leukemia RPMI-8226 and SR
2.2.4. Evaluation of In Vitro Tubulin Polymerization Inhibitory Activity
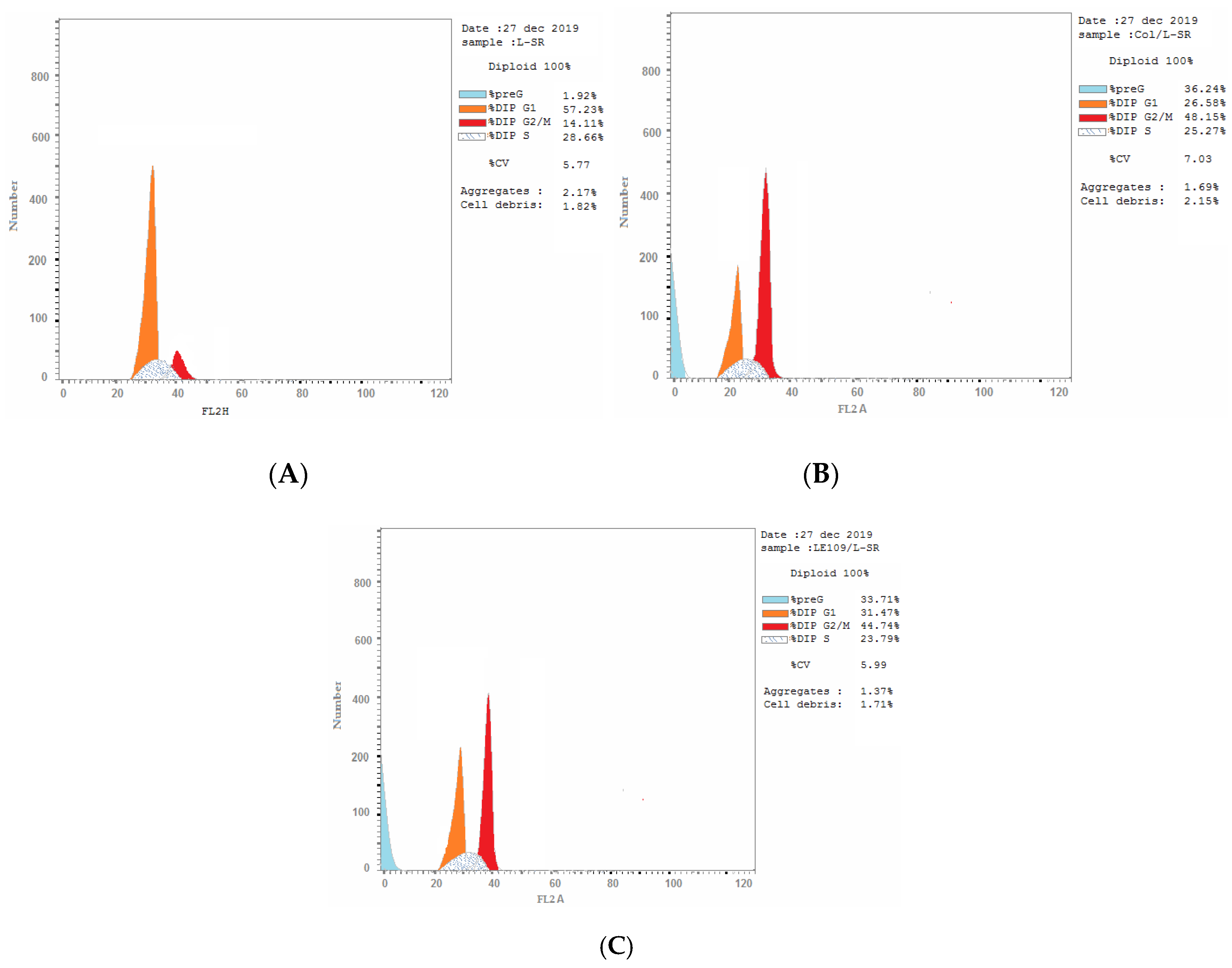
2.2.5. Cell Cycle Analysis
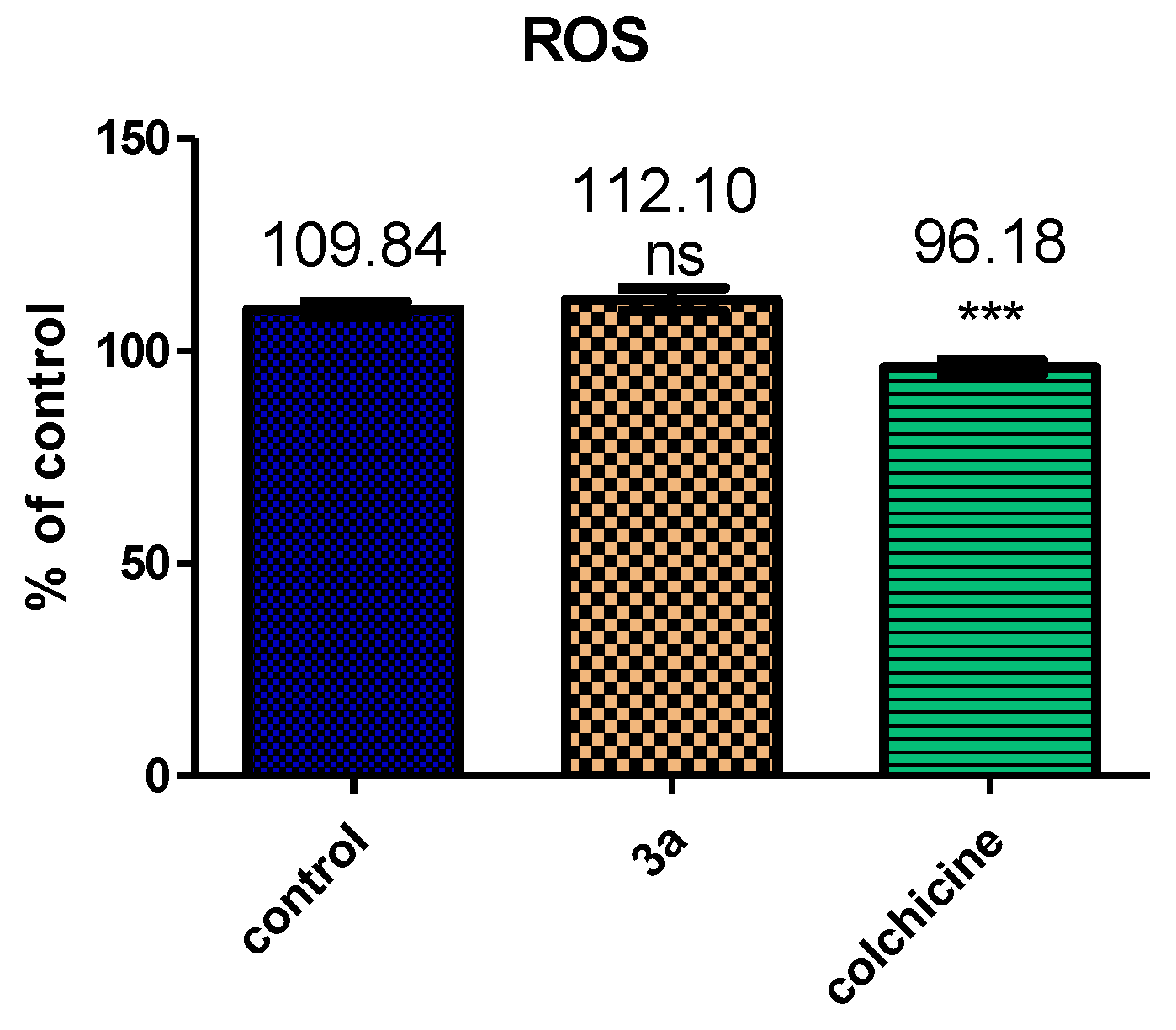
2.2.6. Compound 3b Induced Mitochondrial Depolarization and ROS Production
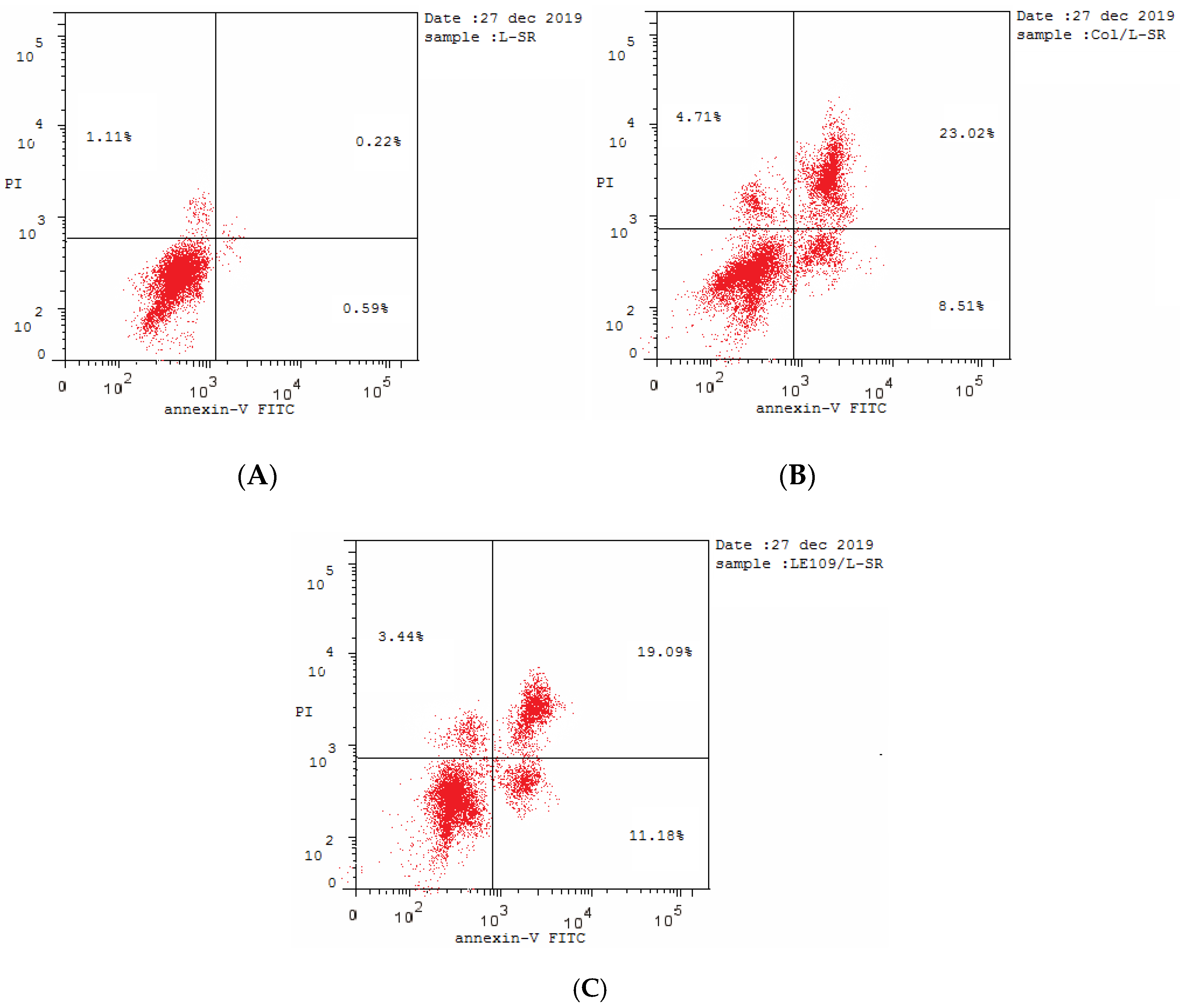
2.2.7. Effect of Compound 3a on Multidrug-Resistant (MDR) Leukemia SR Cells
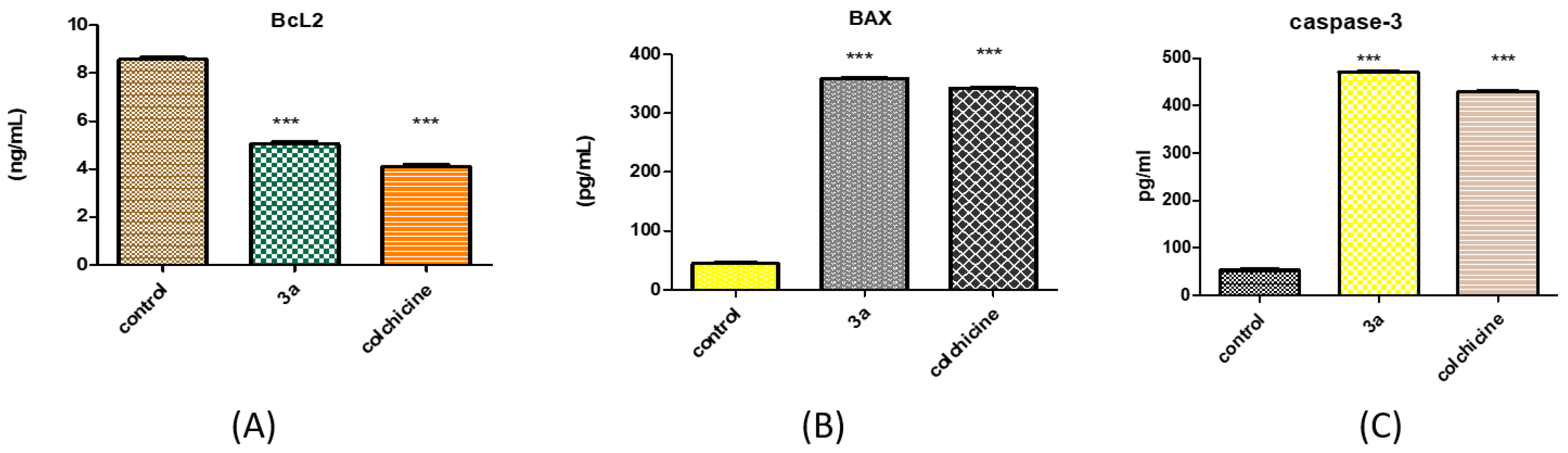
2.2.8. Effect of 3a on Caspase-3 Activation
2.2.9. Effect of 3a on BAX and Bcl-2 Proteins
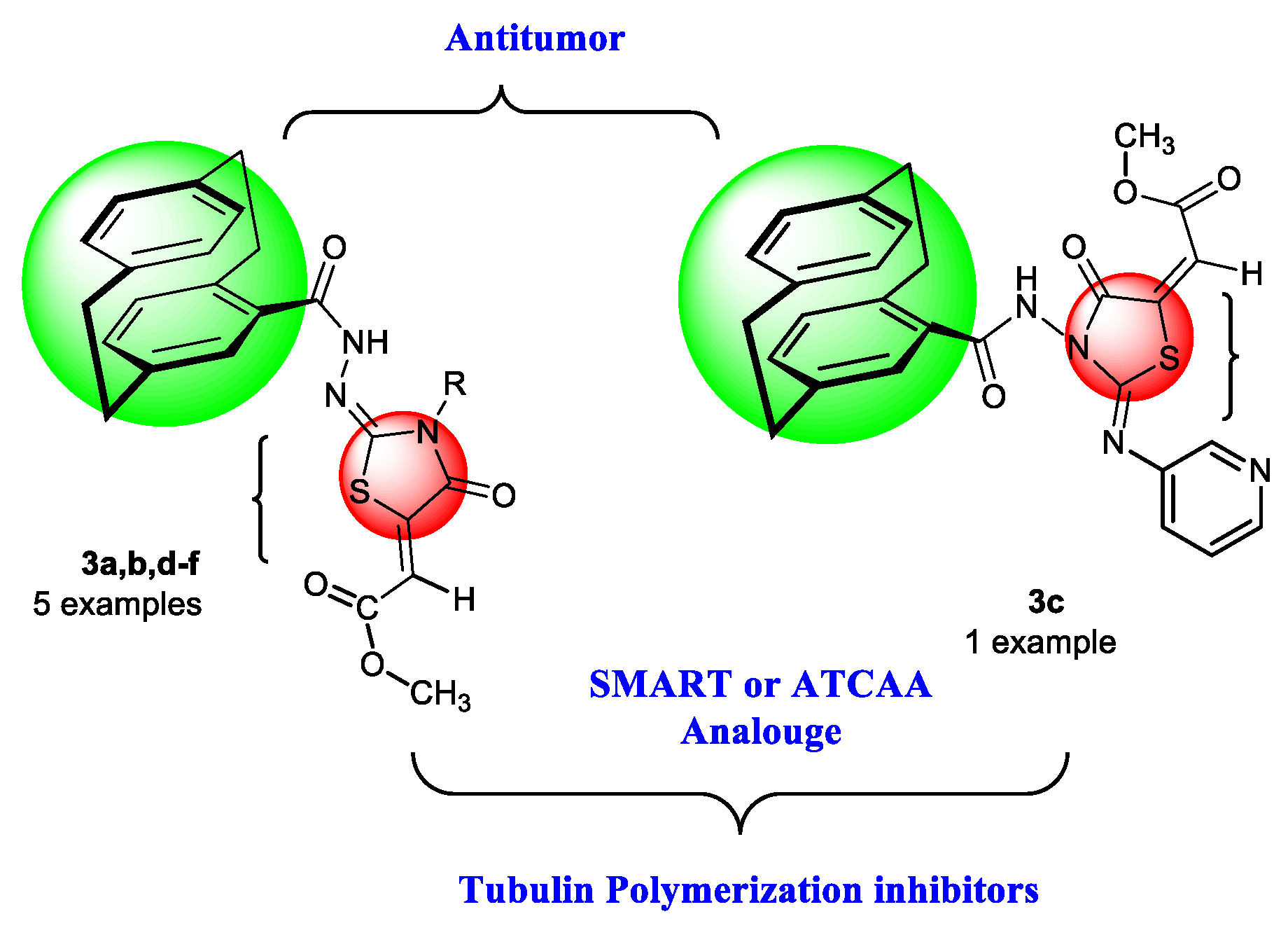
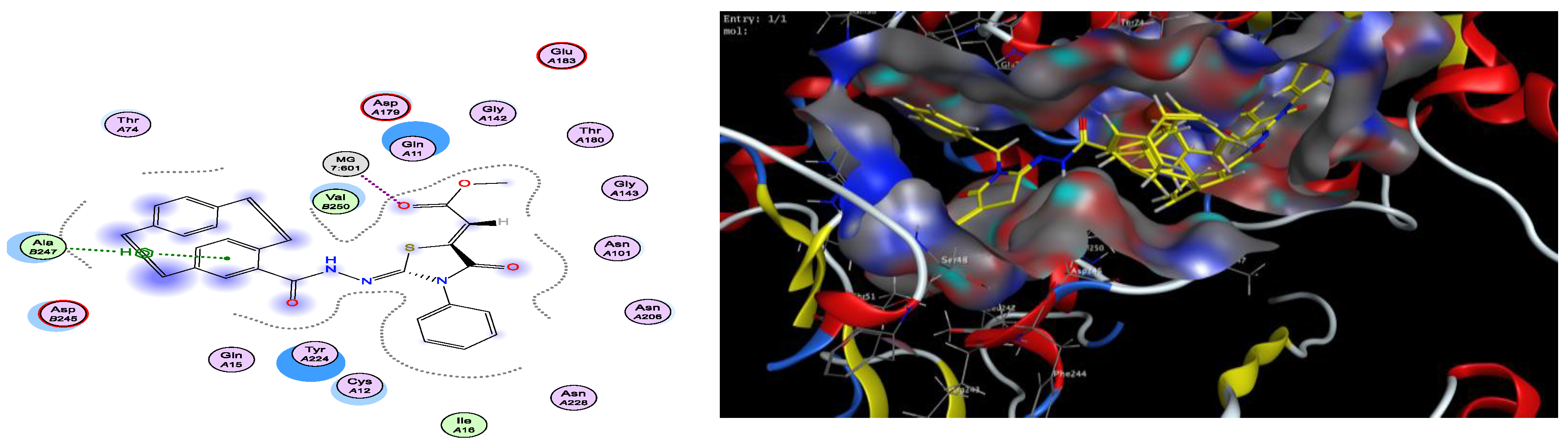
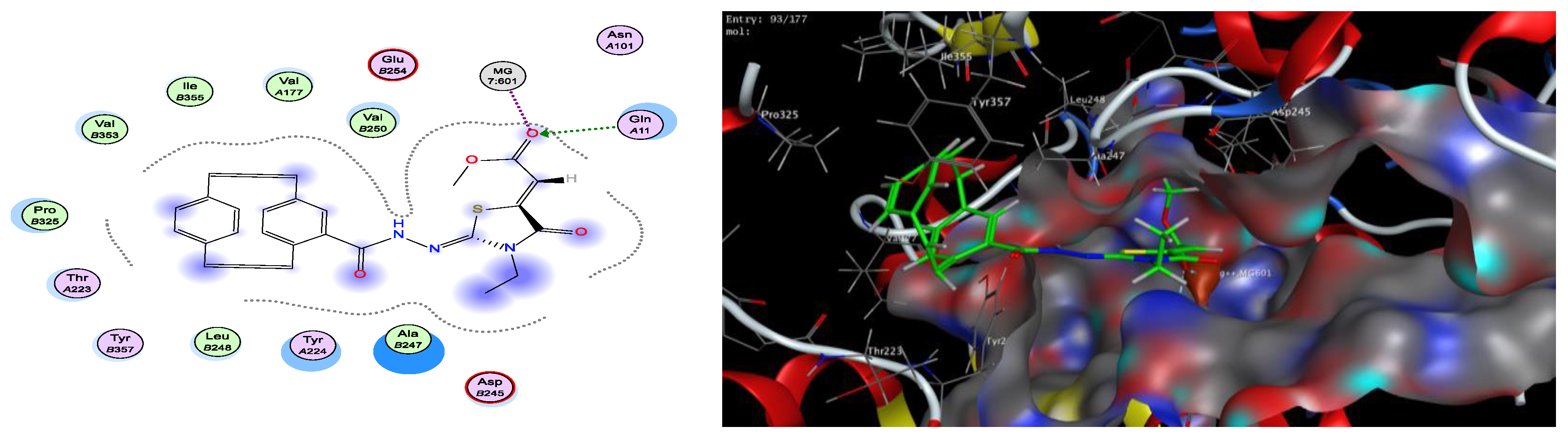
2.3. Docking Studies
3. Experimental
3.1. Chemistry Part
3.1.1. Materials and Methods
3.1.2. Preparation of Paracyclophanyl-N-substituted Hydrazinecarbothioamides 2a–f
3.1.3. Reactions of Hydrazinecarbothioamide Derivatives 2a–f with 9: Preparation of Compounds 3a–f
3.2. Biology Part
NCI Screening Assay
3.3. Docking Studies
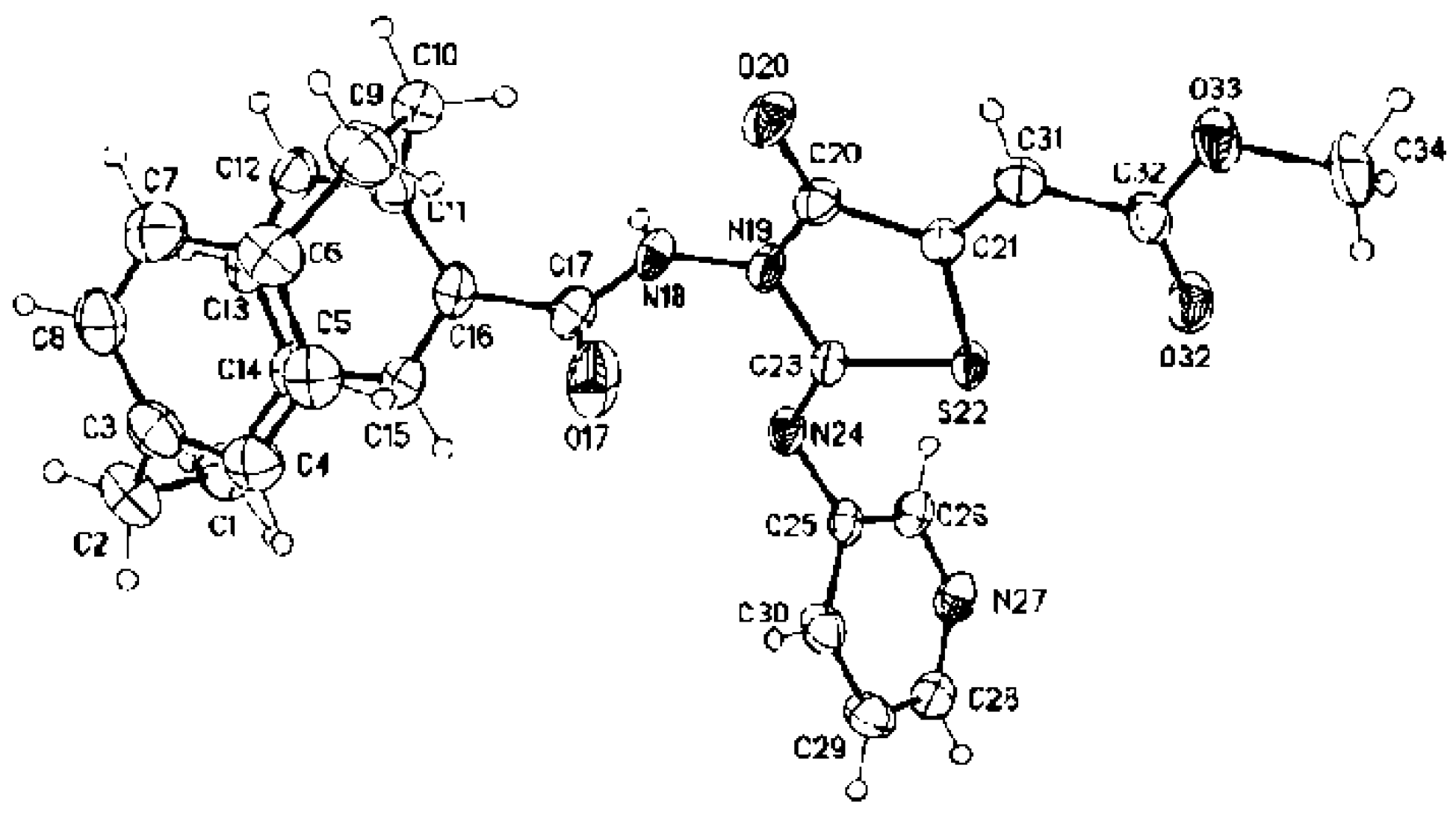
3.4. Crystallographic Structure Analyses
Crystal Structure Determinations of 2a, 2b, 2d, 3b, and 3c
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hopf, H.; Gleiter, R. Modern Cyclophane Chemistry; Wiley-VCH: Weinheim, Germany, 2004; ISBN 978-3-527-30713-5. [Google Scholar]
- Boekelheide, V. Syntheses and properties of the [2n]Cyclophanes. Top. Curr. Chem. 1983, 113, 87–143. [Google Scholar]
- Cram, D.J.; Steinberg, H. Macro Rings. I. Preparation and Spectra of the Paracyclophanes. J. Am. Chem. Soc. 1951, 73, 5691–5704. [Google Scholar]
- Vögtle, F.; Newmann, P. The Synthesis of [2.2]Phanes. Synthesis 1973, 2, 85–103. [Google Scholar] [CrossRef]
- Staab, H.A.; Knaus, G.H.; Henke, H.-E.; Krieger, C. Elektron-Donor-Acceptor-Verbindungen, XXIX. Elektron-Donor-Acceptor-Paracyclophane mit 7,7,8,8-Tetracyanchinodimethan (TCNQ) als Acceptor-Einheit. Chem. Ber. 1983, 116, 2785–2805. [Google Scholar] [CrossRef]
- Hopf, H.; Marquard, C. Strain Release in Aromatic Molecules: The [2n] Cyclophanes. In Strain and its Implications in Organic Chemistry; Springer: Dordrecht, The Netherlands, 1983; pp. 297–332. [Google Scholar]
- Vögtle, F. Cyclophane Chemistry: Synthesis, Structure and Reactions; Wiley: Chichester, UK, 1993; pp. 71–111. [Google Scholar]
- Rozenberg, V.I.; Sergeeve, E.V.; Hopf, H. Modern Cyclophane Chemistry; Gleiter, R., Hopf, H., Eds.; Wiley-VCH: Weinheim, Germany, 2004; Volume 17, pp. 435–462. [Google Scholar]
- Braun, C.; Nieger, M.; Thiel, W.R.; Bräse, S. [2.2]Paracyclophanes with N-Heterocycles as Ligands for Mono-and Dinuclear Ruthenium(II) Complexes. Chem. A Eur. J. 2017, 23, 15474–15483. [Google Scholar] [CrossRef] [PubMed]
- Aly, A.A.; Brown, A.A. Asymmetric and fused heterocycles based on [2.2]paracyclophane. Tetrahedron 2009, 65, 8055–8089. [Google Scholar] [CrossRef]
- Imming, P.; Graf, M.; Tries, S.; Hirschelmann, R.; Krause, E.; Pawlitzki, G. Anti-inflammatory planar chiral [2.2]paracyclophaneacetic acid enantiomers. Inflamm. Res. 2001, 50, 371–374. [Google Scholar] [CrossRef]
- Schlotter, K.; Boeckler, F.; Hübner, H.; Gmeiner, P. Fancy Bioisosteres: Novel Paracyclophane Derivatives As Super-Affinity Dopamine D3 Receptor Antagonists. J. Med. Chem. 2006, 49, 3628–3635. [Google Scholar] [CrossRef]
- Deep, A.; Jain, S.; Sharma, P.C. Synthesis and anti-inflammatory activity of some novel biphenyl-4-carboxylic acid 5-(arylidene)-2-(aryl)-4-oxothiazolidin-3-yl amides. Acta Pol. Pharm. Drug Res. 2010, 67, 63–67. [Google Scholar]
- Salar, U.; Taha, M.; Khan, K.M.; Ismail, N.H.; Imran, S.; Perveen, S.; Gul, S.; Wadood, A. Syntheses of new 3-thiazolyl coumarin derivatives, in vitro α-glucosidase inhibitory activity, and molecular modeling studies. Eur. J. Med. Chem. 2016, 122, 196–204. [Google Scholar] [CrossRef]
- Hassan, A.A.; Mohamed, N.K.; El-Haleem, L.E.A.; Braese, S.; Martin, N. Facile Synthesis of Naphtho [2,3-d]thiazoles, Naphtho[2,3-e][1,3,4]thiadiazines and Bis(naphtho[2,3-d]thiazolyl)-copper(II) Derivatives from Heteroylthiosemicarbazides. Chin. J. Chem. 2016, 34, 814–822. [Google Scholar] [CrossRef]
- Aly, A.A.; Hassan, A.A.; Mohamed, N.K.; El Shaieb, K.; Makhlouf, M.M.; Bräse, S.; Nieger, M. Reactive intermediates in the reaction of hydrazinecarbothioamides with 2-bis-(methylthio)-methylene)malononitrile and ethyl 2-cyano-3,3-bis(methylthio)acrylate. Res. Chem. Intermeds 2019, 45, 613–631. [Google Scholar] [CrossRef] [Green Version]
- Aly, A.A.; Ibrahim, M.A.A.; Shehata, E.M.; Hassan, A.A.M.; Brown, A.B. Prospective new amidinothiazoles as leukotriene B4 inhibitors. J. Mol. Struct. 2019, 1175, 414–427. [Google Scholar] [CrossRef]
- Hassan, A.A.; Aly, A.A.; Mostafa, S.M.; Döpp, D. Formation of thiadiazole, thiadiazine, thiadiazepine and pyrazole derivatives in the reaction of 2,4-disubstituted thiosemicarbazides with tetracyanoethylene. Arkivoc 2018, iii, 200–211. [Google Scholar]
- Aly, A.A.; Hassan, A.A.; AbdAl-Latif, E.-S.M.; Ibrahim, M.A.A.; Bräse, S.; Nieger, M. Reaction of N,N-disubstituted hydrazinecarbothioamides with 2-bromo-2-substituted acetophenones. Arkivoc 2018, iii, 102–111. [Google Scholar]
- Aly, A.A.; Hassan, A.A.; El-Latif, E.-S.M.A. Review on “An update of the use of thiocarbohydrazides and thiosemicarbazides in the preparation of heterocycles and their biological importance”. J. Heterocycl. Chem. 2018, 55, 2196–2223. [Google Scholar] [CrossRef]
- Aly, A.A.; Ishak, E.A.; El Malah, T.; Brown, A.B.; Elayah, W.M. Synthesis of potentially antioxidant and antibacterial biologically active thiazolidines. J. Heterocycl. Chem. 2015, 52, 1758–1764. [Google Scholar] [CrossRef]
- Aly, A.A.; Mohamed, N.K.; Hassan, A.A.; El-Shaieb, K.M.; Makhlouf, M.M.; Bräse, S.; Nieger, M.; Brown, A.B. Functionalized 1,3-Thiazolidin-4-Ones from 2-Oxo-Acenaphtho-quinylidene-and [2.2]Paracyclophanylidene-Thiosemicarbazones. Molecules 2019, 24, 3069. [Google Scholar] [CrossRef] [Green Version]
- Aly, A.A.; Mohamed, A.H.; Ramadan, M. Synthesis and colon anticancer activity of some novel thiazole/-2-quinolone derivatives. J. Mol. Struct. 2020, 1207, in press. [Google Scholar] [CrossRef]
- Aly, A.A.; Bräse, S.; Weis, P. Tridentate and bidentate copper complexes of [2.2]paracyclophanyl-substituted thiosemicarbazones, thiocarbazones, hydrazones and thioureas. J. Mol. Struct. 2019, 1178, 311–326. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Z.; Li, C.-M.; Lu, Y.; Vaddady, P.K.; Meibohm, B.; Dalton, J.T.; Miller, D.D.; Li, W. Discovery of novel 2-aryl-4-benzoyl-imidazoles targeting the colchicines binding site in tubulin as potential anticancer agents. J. Med. Chem. 2010, 53, 7414–7427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bestgen, S.; Seidl, C.; Wiesner, T.; Zimmer, A.; Falk, M.; Köberle, B.; Austeri, M.; Paradies, J.; Bräse, S.; Schepers, U. Double-Strand DNA Breaks Induced by Paracyclophane Gold (I) Complexes. Chem. A Eur. J. 2017, 23, 6315–6322. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-M.; Wang, Z.; Lu, Y.; Ahn, S.; Narayanan, R.; Kearbey, J.D.; Parke, D.N.; Li, W.; Mille, D.D.; Dalton, J.T. Biological activity of 4-substituted methoxybenzoyl-aryl-thiazole: An active microtubule inhibitor. Cancer Res. 2011, 71, 216–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagnoli, R.; Baraldi, P.G.; Salvador, M.K.; Preti, D.; Tabrizi, M.A.; Brancale, A.; Fu, X.-H.; Li, J.; Zhang, S.-Z.; Hamel, E. Discovery and optimization of a series of 2-aryl-4-amino-5-(3′,4′,5′-trimethoxybenzoyl)thiazoles as novel anticancer agents. J. Med. Chem. 2012, 55, 5433–5445. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, C.-M.; Wang, Z.; Ross, C.R.; Chen, J.; Dalton, J.T.; Li, W.; Miller, D.D. Discovery of 4-substituted methoxybenzoyl-aryl-thiazole as Novel Anticancer Agents: Synthesis, biological evaluation, and structure−activity relationships. J. Med. Chem. 2009, 52, 1701–1711. [Google Scholar] [CrossRef] [Green Version]
- Aly, A.A.; Bräse, S.; Hassan, A.A.; Mohamed, N.K.; El-Haleem, L.E.A. New planar-chiral linked [2.2]paracyclophanes-N-([2.2]-paracyclophanylcarbamoyl)-4-([2.2]paracyclophanylcarboxamide. Molecules 2020. under review. [Google Scholar]
- Zitt, H.; Dix, I.; Hopf, H.; Jones, P.G. 4,15-Diamino[2.2]paracyclophane, a Reusable Template for Topochemical Reaction Control in Solution. Eur. J. Org. Chem. 2002, 14, 2298–2307. [Google Scholar] [CrossRef]
- Holbeck, S.L.; Camalier, R.; Crowell, J.A.; Govindharajulu, J.P.; Hollingshead, M.; Anderson, L.W.; Polley, E.; Rubinstein, L.; Srivastava, A.; Wilsker, D. The National Cancer Institute ALMANAC: A comprehensive screening resource for the detection of anticancer drug pairs with enhanced therapeutic activity. Cancer Res. 2017, 77, 3564–3576. [Google Scholar] [CrossRef] [Green Version]
- Rostom, S.A. Synthesis and in vitro antitumor evaluation of some indeno[1,2-c]pyrazol (in) es substituted with sulfonamide, sulfonylurea (-thiourea) pharmacophores, and some derived thiazole ring systems. Biorg. Chem. Med. Chem. 2006, 14, 6475–6485. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef]
- Nohl, H.; Gille, L.; Staniek, K. Intracellular generation of reactive oxygen species by mitochondria. Biochem. Pharmacol. 2005, 69, 719–723. [Google Scholar] [CrossRef]
- Rae, M.; Creighton, C.J.; Meck, J.M.; Haddad, B.R.; Johnson, M.D. MDA-MB-435 cells are derived from M14 Melanoma cells––a loss for breast cancer, but a boon for melanoma research. Breast Cancer Res. Treat. 2007, 104, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Polgar, O.; Bates, S.E. ABC transporters in the balance: Is there a role in multidrug resistance? Biochem. Soc. Trans. 2005, 33, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Caspase function in programmed cell death. Cell Death Differ. 2007, 14, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Matson, D.R.; Stukenberg, P.T. Spindle poisons and cell fate: A tale of two pathways. Mol. Interv. 2011, 11, 141–149. [Google Scholar] [CrossRef]
- Clarke, P.R.; Allan, L.A. Cell-cycle control in the face of damage–a matter of life or death. Trends Cell Boil. 2009, 19, 89–98. [Google Scholar] [CrossRef]
- Tolosa, L.; Donato, M.T.; Gomez-Lecchon, M.J. General Cytotoxicity Assessment by Means of the MTT Assay. Methods Mol. Biol. 2015, 1250, 333–348. [Google Scholar]
- Pozarowski, P.; Darzylienwicz, Z. Analysis of cell cycle by flow cytometry. Methods Mol. Biol. 2004, 281, 301–311. [Google Scholar]
- Niles, A.L.; Moravec, R.A.; Riss, T.L. Caspase activity assays. Methods Mol. Biol. 2008, 414, 137–150. [Google Scholar] [PubMed]
- Peng, L.X.; Wallace, M.; Andaloro, B.; Fallon, D.; L, L.F.; Delduco, D.; Tice, G. Modification of the BAX System PCR assay for detecting Salmonella in beef, produce, and soy protein isolate. Performance Tested Method 100201. J. AOAC Int. 2011, 94, 172–178. [Google Scholar] [CrossRef] [Green Version]
- Redmann, M.; Benavides, G.A.; Wani, W.Y.; Berryhill, T.F.; Ouyang, X.; Johnson, M.S.; Ravi, S.; Mitra, K.; Barnes, S.; Darley-Usmar, V.M.; et al. Methods for assessing mitochondrial quality control mechanisms and cellular consequences in cell culture. Redox Biol. 2018, 17, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Mille, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Yotnda, P. Production and detection of reactive oxygen species (ROS) in cancers. J Vis. Exp. 2011, 57, 3357–3365. [Google Scholar] [CrossRef] [PubMed]
- Gilpin, C.; Korobitsyn, A.; Weyer, K. Current tools available for the diagnosis of drug-resistant tuberculosis. Ther. Adv. Infect. Dis. 2016, 3, 145–151. [Google Scholar] [CrossRef] [PubMed]
- OpenEye Scientific Software Fast Rigid Exhaustive Docking (FRED) Receptor, Version 2.2.5. Available online: http://www.eyesopen.com (accessed on 27 May 2020).
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal strturcture refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. 2009, D65, 148–155. [Google Scholar] [CrossRef]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Panel/Cell Line | 3a | 3b | 3c | 3d | 3e | 3f | |
|---|---|---|---|---|---|---|---|
| Leukemia | CCRF-CEM | 87.04 | 96.17 | 31.27 | 77.08 | 66.08 | 41.43 |
| HL-60(TB) | 83.60 | 105.46 | 48.64 | 8.16 | 11.64 | 26.38 | |
| K-562 | 72.95 | 81.13 | 20.72 | 48.13 | 45.78 | 30.52 | |
| MOLT-4 | 96.97 | 98.91 | 21.86 | 64.55 | 62.85 | 55.61 | |
| RPMI-8226 | 120.89 | 147.00 | 83.01 | 109.36 | 114.28 | 49.83 | |
| SR | 115.60 | 114.70 | 74.18 | 98.21 | 113.40 | 39.13 | |
| Non-Small-Cell Lung Cancer | A549/ATCC | 30.92 | 22.95 | 2.04 | 5.20 | 3.00 | 11.39 |
| EKVX | 41.03 | 22.30 | 2.61 | 9.16 | 6.44 | 12.16 | |
| HOP-62 | 97.00 | 22.76 | 4.00 | 0 | 0 | 0 | |
| HOP-92 | 24.66 | 24.50 | 8.14 | 18.59 | 18.43 | 16.16 | |
| NCI-H226 | 26.22 | 35.20 | 7.48 | 12.45 | 7.31 | 9.41 | |
| NCI-H23 | 59.57 | 22.64 | 9.18 | 11.85 | 0.40 | 9.32 | |
| NCI-H322M | 31.54 | 6.70 | 1.72 | 2.94 | 7.75 | 4.43 | |
| NCI-H460 | 39.35 | 6.29 | 0.75 | 0 | 0 | 1.98 | |
| NCI-H522 | 58.47 | 84.06 | 17.74 | 39.25 | 40.31 | 30.01 | |
| Colon Cancer | COLO 205 | 52.26 | 27.31 | 0 | 0 | 0 | 4.48 |
| HCC-2998 | 96.36 | 20.49 | 0 | 0 | 0 | 0 | |
| HCT-116 | 95.95 | 50.48 | 6.35 | 46.02 | 50.54 | 24.59 | |
| HCT-15 | 99.66 | 72.16 | 11.75 | 33.11 | 31.60 | 29.43 | |
| HT29 | 113.13 | 87.62 | 7.71 | 53.99 | 54.76 | 23.99 | |
| KM12 | 91.51 | 34.90 | 7.32 | 18.40 | 15.01 | 1.32 | |
| SW-620 | 92.61 | 81.83 | 10.84 | 55.46 | 91.21 | 12.94 | |
| CNS Cancer | SF-268 | 27.49 | 14.95 | 3.08 | 0 | 2.18 | 2.56 |
| SF-295 | 41.00 | 10.63 | 5.63 | 6.42 | 3.13 | 8.80 | |
| SF-539 | 122.50 | 56.74 | 11.63 | 0.33 | 9.82 | 0 | |
| SNB-19 | 60.06 | 43.08 | 16.64 | 15.31 | 14.43 | 11.26 | |
| SNB-75 | 54.29 | 48.78 | 0.25 | 21.92 | 21.18 | 24.73 | |
| U251 | 84.20 | 45.04 | 14.66 | 18.17 | 15.64 | 20.27 | |
| Melanoma | LOX IMVI | 127.77 | 93.79 | 9.78 | 21.22 | 26.51 | 9.41 |
| MALME-3M | 21.23 | 11.57 | 0 | 7.56 | 8.06 | 11.28 | |
| M14 | 46.88 | 36.59 | 7.99 | 21.14 | 12.85 | 19.23 | |
| MDA-MB-435 | 10.39 | 8.44 | 0 | 0 | 0.72 | 8.06 | |
| SK-MEL-2 | 12.98 | 14.42 | 0 | 2.70 | 5.78 | 13.53 | |
| SK-MEL-28 | 17.54 | 0 | 0.86 | 0 | 0 | 10.28 | |
| SK-MEL-5 | 30.65 | 17.72 | 3.64 | 7.53 | 7.50 | 14.50 | |
| UACC-257 | 30.75 | 19.02 | 0.83 | 8.89 | 7.26 | 16.33 | |
| UACC-62 | 74.85 | 48.11 | 10.67 | 22.84 | 21.39 | 17.97 | |
| Ovarian Cancer | IGROV1 | 94.03 | 57.72 | 15.44 | 10.76 | 11.62 | 7.20 |
| OVCAR-3 | 106.25 | 79.71 | 4.51 | 13.56 | 7.09 | 15.87 | |
| OVCAR-4 | 70.61 | 55.43 | 7.76 | 2.93 | 10.00 | 10.51 | |
| OVCAR-5 | 51.75 | 34.65 | 0.61 | 3.90 | 0 | 0 | |
| OVCAR-8 | 59.27 | 28.00 | 1.51 | 11.78 | 7.42 | 13.28 | |
| NCI/ADR-RES | 68.61 | 29.61 | 4.70 | 10.50 | 13.46 | 7.72 | |
| SK-OV-3 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Renal Cancer | 786-0 | 100.28 | 50.45 | 8.52 | 8.77 | 9.45 | 14.62 |
| A498 | 50.75 | 53.92 | 3.59 | 12.10 | 6.63 | 3.39 | |
| ACHN | 30.67 | 34.18 | 11.02 | 12.21 | 10.45 | 17.78 | |
| CAKI-1 | 94.65 | 84.43 | 4.78 | 5.23 | 0 | 24.49 | |
| SN12C | 77.51 | 38.76 | 8.87 | 13.14 | 4.97 | 17.07 | |
| TK-10 | 84.83 | 4.53 | 0 | 0 | 6.50 | 0 | |
| UO-31 | 41.51 | 54.96 | 21.86 | 24.62 | 21.03 | 21.73 | |
| Prostate Cancer | PC-3 | 135.88 | 82.23 | 18.19 | 51.62 | 43.28 | 18.95 |
| DU-145 | 49.13 | 16.79 | 0 | 0 | 0 | 6.20 | |
| Breast Cancer | MCF7 | 76.79 | 67.26 | 26.75 | 48.47 | 56.29 | 36.38 |
| MDA-MB-231/ATCC | 67.47 | 43.16 | 10.64 | 2.27 | 3.95 | 3.50 | |
| HS 578T | 22.06 | 12.58 | 0 | 6.41 | 1.03 | 5.01 | |
| BT-549 | 64.50 | 89.68 | 18.41 | 38.28 | 7.73 | 26.81 | |
| T-47D | 138.25 | 106.25 | 31.13 | 25.00 | 5.77 | 23.00 | |
| MDA-MB-468 | 100.39 | 78.91 | 18.03 | 23.68 | 12.74 | 29.74 | |
| Panel | Cell Line | GI50 | TGI | LC50 | ||||
|---|---|---|---|---|---|---|---|---|
| Concentration/Cell Line | Subpanel MIDb | Selectivity Ratio (MIDa: MIDb) | Concentration/Cell Line | Subpanel MIDd | Selectivity Ratio (MIDc: MIDd) | |||
| Leukemia | CCRF-CEM | 2.71 | 2.72 | 0.88 | - | 81.14 | 3.85 | >100 |
| HL-60(TB) | 2.70 | >100 | >100 | |||||
| K-562 | 3.15 | >100 | >100 | |||||
| MOLT-4 | 3.12 | >100 | >100 | |||||
| RPMI-8226 | 2.15 | 5.69 | >100 | |||||
| SR | 2.46 | >100 | >100 | |||||
| Non-Small-Cell Lung Cancer | A549/ATCC | 2.83 | 2.74 | 0.87 | 8.00 | 16.05 | 1.31 | >100 |
| EKVX | 1.91 | 4.23 | 9.38 | |||||
| HOP-62 | 2.68 | 6.97 | 6.60 | |||||
| HOP-92 | 1.91 | 9.14 | >100 | |||||
| NCI-H226 | 4.38 | 2.55 | >100 | |||||
| NCI-H23 | 1.95 | 4.15 | 8.85 | |||||
| NCI-H322M | 2.02 | 4.75 | 1.32 | |||||
| NCI-H460 | 2.08 | 4.29 | 8.82 | |||||
| NCI-H522 | 4.89 | >100 | >100 | |||||
| Colon Cancer | COLO 205 | 2.13 | 1.91 | 1.24 | 4.61 | 3.95 | 5.34 | |
| HCC-2998 | 1.78 | 3.45 | 6.68 | |||||
| HCT-116 | 1.79 | 3.66 | 7.51 | |||||
| HCT-15 | 1.87 | 4.05 | 8.76 | |||||
| HT29 | 1.93 | 4.07 | 8.56 | |||||
| KM12 | 2.03 | 4.15 | 8.51 | |||||
| SW-620 | 1.85 | 3.66 | 7.23 | |||||
| CNS Cancer | SF-268 | 3.09 | 2.71 | 0.88 | 1.12 | 19.48 | 1.08 | >100 |
| SF-295 | 2.07 | 4.19 | 8.50 | |||||
| SF-539 | 1.79 | 3.32 | 6.18 | |||||
| SNB-19 | 1.95 | 4.25 | 9.26 | |||||
| SNB-75 | 5.44 | >100 | >100 | |||||
| U251 | 1.89 | 4.00 | 8.47 | |||||
| Melanoma | LOX IMVI | 1.69 | 3.77 | 0.63 | 3. | 36.54 | 0.58 | 6.75 |
| MALME-3M | 3.27 | 2.14 | >100 | |||||
| M14 | 3.27 | 1.54 | >100 | |||||
| MDA-MB-435 | 5.44 | 100 | >100 | |||||
| SK-MEL-2 | 2.65 | 7.76 | >100 | |||||
| SK-MEL-28 | 2.97 | 9.87 | >100 | |||||
| SK-MEL-5 | 7.56 | >100 | >100 | |||||
| UACC-257 | 5.14 | >100 | >100 | |||||
| UACC-62 | 1.96 | 4.21 | 9.02 | |||||
| Ovarian Cancer | IGROV1 | 1.66 | 2.59 | 0.92 | 3.27 | 18.46 | 1.14 | 6.44 |
| OVCAR-3 | 1.80 | 3.61 | 7.23 | |||||
| OVCAR-4 | 3.17 | 9.88 | 3.69 | |||||
| OVCAR-5 | 1.92 | 3.84 | 7.68 | |||||
| OVCAR-8 | 3.08 | >100 | >100 | |||||
| NCI/ADR-RES | 2.92 | 1.06 | >100 | |||||
| SK-OV-3 | 3.55 | 7.53 | >100 | |||||
| Renal Cancer | 786-0 | 2.00 | 1.86 | 1.28 | 4.38 | 16.64 | 1.27 | 9.58 |
| A498 | -- | 8.36 | >100 | |||||
| ACHN | 1.78 | 3.29 | 6.08 | |||||
| RXF393 | 3.74 | 3.86 | >100 | |||||
| CAKI-1 | 1.52 | 3.00 | 9.78 | |||||
| SN12C | 2.27 | 5.94 | >100 | |||||
| TK-10 | 2.03 | 5.60 | 2.92 | |||||
| UO-31 | 1.53 | 3.08 | 6.20 | |||||
| Prostate Cancer | PC-3 | 1.71 | 3.41 | 0.69 | 3.63 | 3.58 | 5.89 | 7.70 |
| DU-145 | 5.11 | 3.53 | >100 | |||||
| Breast Cancer | MCF7 | 1.87 | 2.27 | 1.05 | 4.95 | 24.28 | 0.87 | 2.25 |
| MDA-MB-231/ATCC | 2.63 | 7.46 | >100 | |||||
| HS 578T | 3.49 | >100 | >100 | |||||
| BT-549 | 2.22 | 5.35 | 2.47 | |||||
| T-47D | 1.54 | 3.65 | 8.68 | |||||
| MDA-MB-468 | 1.84 | 3.74 | 7.61 | |||||
| MIDa | 2.38 | MIDc | 21.08 | |||||
| Compound | Cytotoxicity IC50 (µM) a | |
|---|---|---|
| RPMI-8226 | L.SR | |
| 3a | 1.61 ± 0.0654 *** | 1.11 ± 0.06849 *** |
| 3b | 4.62 ± 0.0459 *** | 2.02 ± 0.06875 *** |
| 3c | 9.96 ± 0.3451 *** | 4.84 ± 0.08425 *** |
| 3d | 17.81 ± 0.0544 *** | 22.06 ± 0.06792 *** |
| 3e | 3.17 ± 0.0489 *** | 5.04 ± 0.07908 *** |
| Colchicine | 4.05 ± 0.0478 *** | 1.81 ± 0.07009 *** |
| Compound | Tubulin Polymerization Inhibition IC50 (µM) a |
|---|---|
| 3a | 4.97 ± 0.11 |
| 3b | 8.38 ± 0.18 |
| 3c | 8.28 ± 0.18 |
| 3d | 14.79 ± 0.32 |
| 3e | 6.61 ± 0.14 |
| Colchicine | 3.76 ± 0.08 |
| Compound | DNA Content % | ||||
|---|---|---|---|---|---|
| %G0-G1 | %S | %G2-M | %Pre G1 | Comment | |
| 3a/SR | 31.47 | 23.79 | 44.74 ± 0.1747 *** | 33.71 | cell growth arrest (G2/M) |
| Colchicine/SR | 26.58 | 25.27 | 48.15 ± 1.000 *** | 36.24 | cell growth arrest (G2/M) |
| cont.SR | 57.23 | 28.66 | 14.11 ± 0.1050 | 1.92 | --- |
| Compound Code | IC50 ± SEM (ng/mL) (n = 3) | |
|---|---|---|
| Pgp-Mediated MDR | Fold Change | |
| 3a/SR | 1.285 ± 0.06 | 1.15 |
| Colchicine/SR | 1.726 ± 0.05 | 1.54 |
| Control/SR | 1.121 ± 0.05 | 1 |
| Compound | Caspase 3 | |
|---|---|---|
| Conc pg/mL | Fold Change | |
| 3a/SR | 471.2 ± 6.11 | 8.84 |
| Colchicine/SR | 428.9 ± 5.47 | 8.05 |
| Control/SR | 53.28 ± 1.09 | 1 |
| Compound | BAX | Bcl2 | ||
|---|---|---|---|---|
| Concentration (pg/mL) | Fold Change | Concentration (ng/mL) | Fold Change | |
| 3a/SR | 359 ± 3.830 *** | 7.98 | 5.045 ± 0.1918 *** | 0.59 |
| Colchicine/SR | 341.8 ± 4.310 *** | 7.60 | 4.101 ± 0.1441 *** | 0.48 |
| Control/SR | 44.96 ± 1.270 | 1 | 8.569 ± 0.1565 | 1 |
| Compound | S Score | ΔG (kcal/mol)a | Ligand–Receptor Interaction | ||
|---|---|---|---|---|---|
| Residue | Type | Length (Å) | |||
| Colchicine | −5.62 | −3.4 −0.5 | Glu 71 Mg 601 | H–donor metal | 3.16 2.61 |
| 3a | −7.05 | −2.2 −0.6 | Mg 601 Ala 247 | metal pi–H | 2.00 4.17 |
| 3b | −7.16 | −0.9 −2.3 −0.9 | Asp 245 Arg 2 Glu 71 | H–donor H–acceptor pi–H | 3.27 3.41 3.98 |
| 3c | −6.29 | −1.8 −0.7 −0.7 | Mg 601 Tyr 224 Ala 247 | metal pi–H pi–H | 2.40 3.79 4.50 |
| 3d | −6.94 | −1.0 −2.1 −0.7 | Glu 71 Mg 601 Arg 2 | H–donor metal pi–H | 3.90 2.51 4.69 |
| 3e | −6.43 | −2.0 −2.2 | Gln 11 Mg 601 | H–acceptor metal | 3.09 2.33 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aly, A.A.; Bräse, S.; Hassan, A.A.; Mohamed, N.K.; Abd El-Haleem, L.E.; Nieger, M.; Morsy, N.M.; Abdelhafez, E.M.N. New Paracyclophanylthiazoles with Anti-Leukemia Activity: Design, Synthesis, Molecular Docking, and Mechanistic Studies. Molecules 2020, 25, 3089. https://doi.org/10.3390/molecules25133089
Aly AA, Bräse S, Hassan AA, Mohamed NK, Abd El-Haleem LE, Nieger M, Morsy NM, Abdelhafez EMN. New Paracyclophanylthiazoles with Anti-Leukemia Activity: Design, Synthesis, Molecular Docking, and Mechanistic Studies. Molecules. 2020; 25(13):3089. https://doi.org/10.3390/molecules25133089
Chicago/Turabian StyleAly, Ashraf A, Stefan Bräse, Alaa A. Hassan, Nasr K. Mohamed, Lamiaa E. Abd El-Haleem, Martin Nieger, Nesrin M. Morsy, and Elshimaa M. N. Abdelhafez. 2020. "New Paracyclophanylthiazoles with Anti-Leukemia Activity: Design, Synthesis, Molecular Docking, and Mechanistic Studies" Molecules 25, no. 13: 3089. https://doi.org/10.3390/molecules25133089
APA StyleAly, A. A., Bräse, S., Hassan, A. A., Mohamed, N. K., Abd El-Haleem, L. E., Nieger, M., Morsy, N. M., & Abdelhafez, E. M. N. (2020). New Paracyclophanylthiazoles with Anti-Leukemia Activity: Design, Synthesis, Molecular Docking, and Mechanistic Studies. Molecules, 25(13), 3089. https://doi.org/10.3390/molecules25133089