High-Throughput Omics and Statistical Learning Integration for the Discovery and Validation of Novel Diagnostic Signatures in Colorectal Cancer

 ,
,
Abstract
:
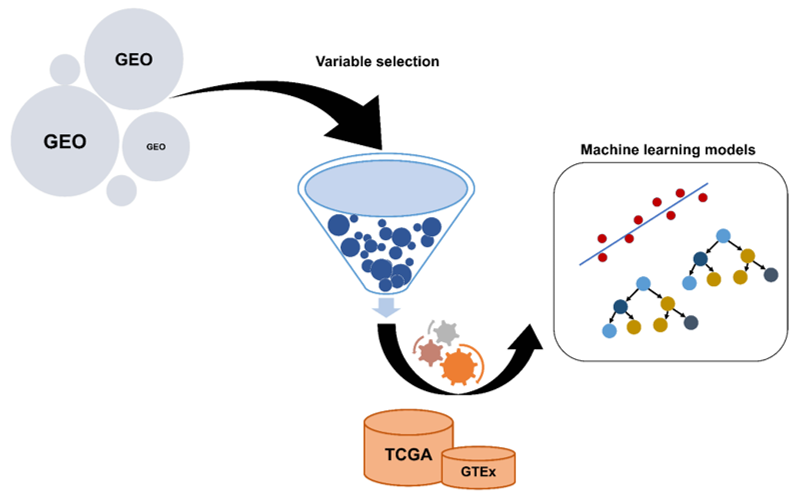
1. Introduction
2. Results
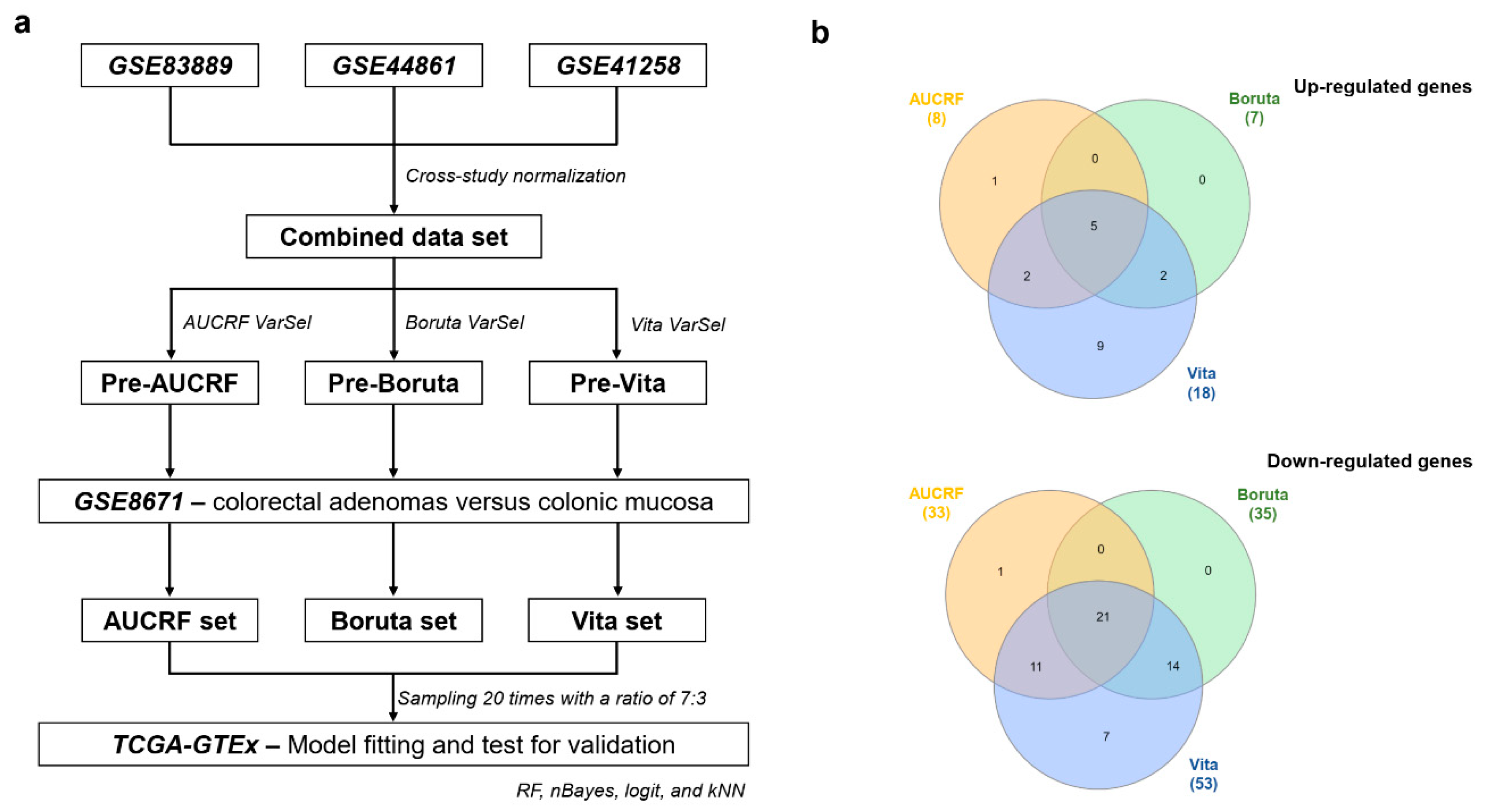
2.1. The Construction of Genetic Biosignatures for the Diagnosis of Colorectal Cancer
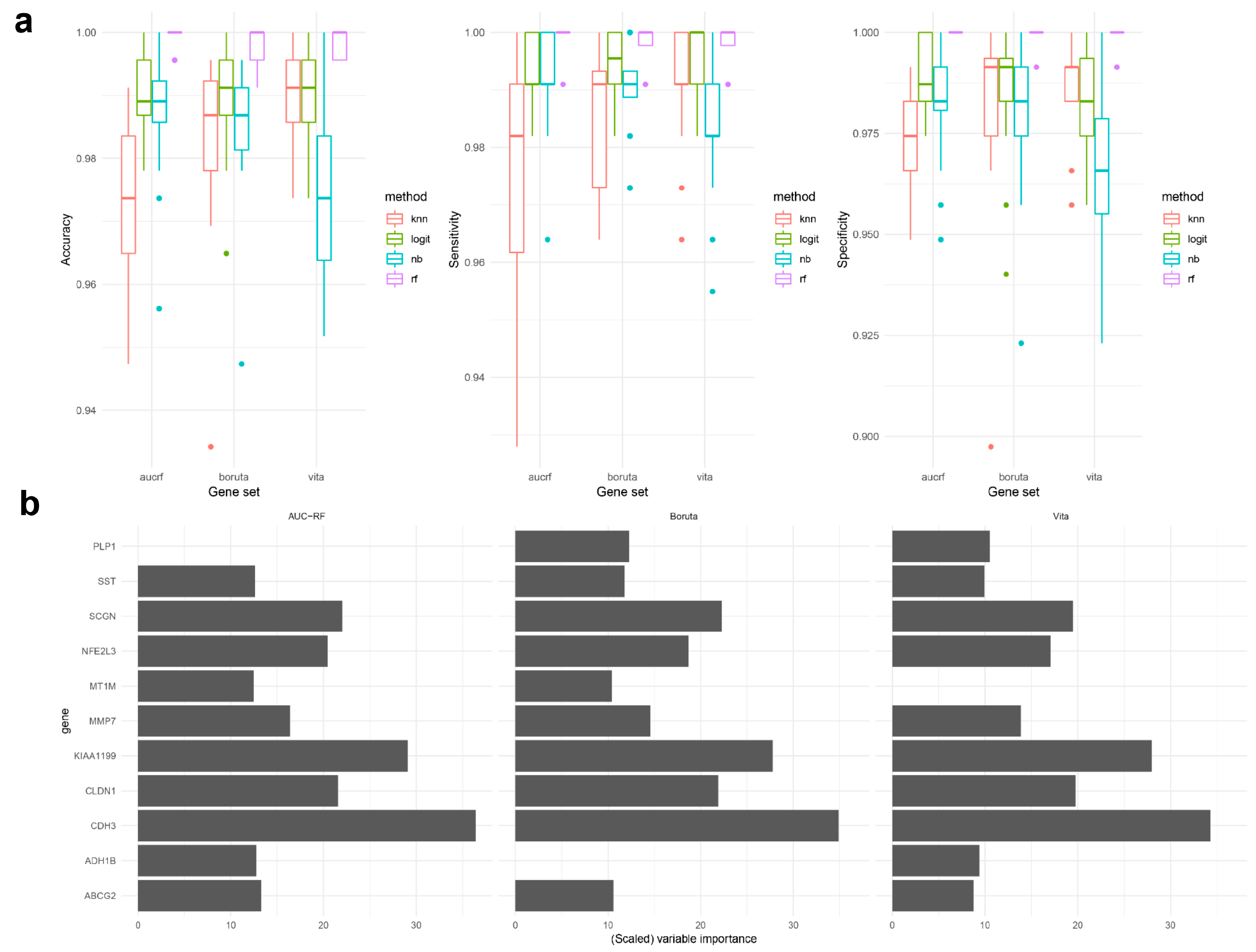
2.2. Classification Model Development, Validation, and Comparisons of the Three Genetic Biosignatures
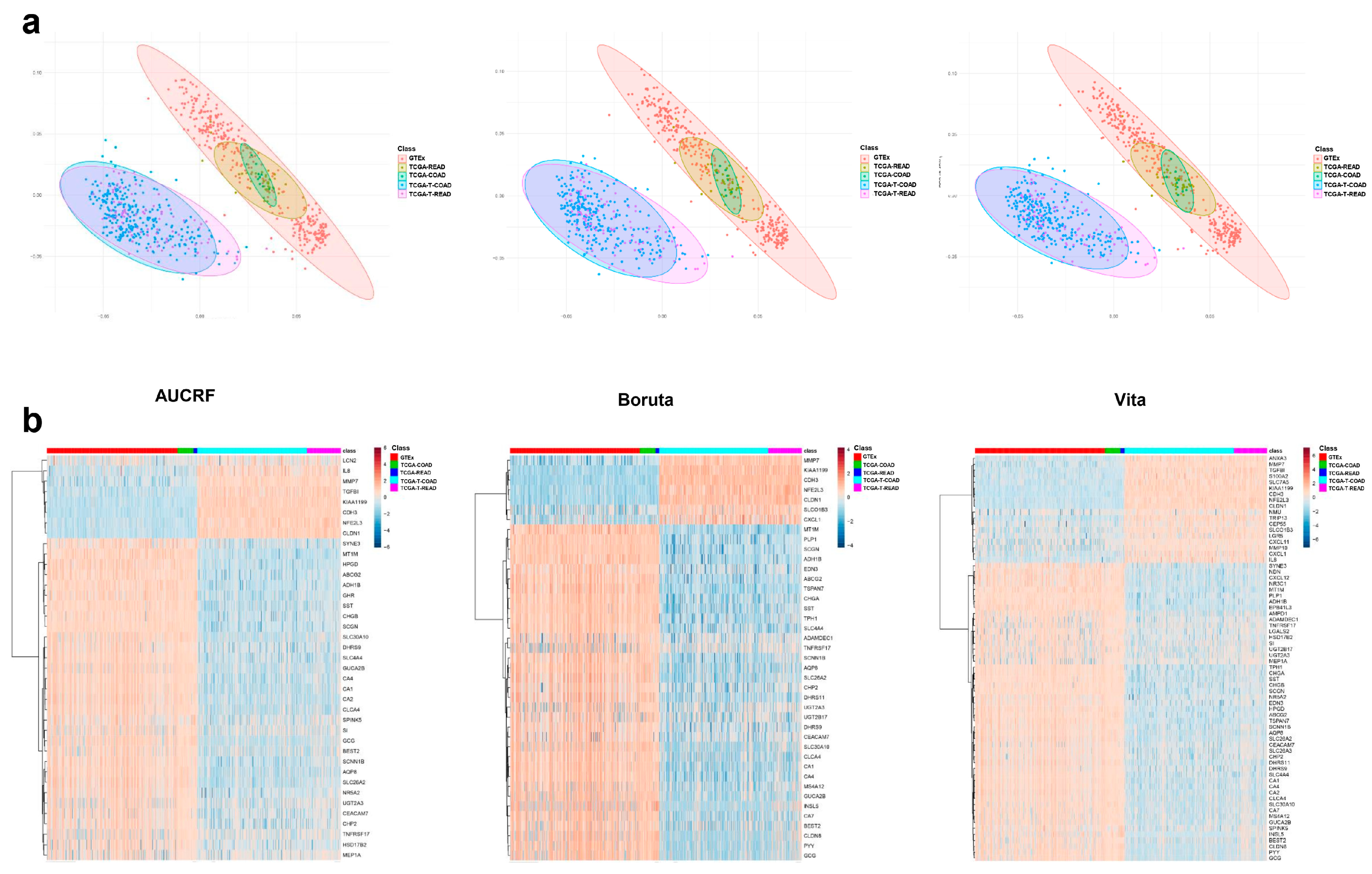
2.2.1. Data Exploration and Visualization
2.2.2. Model Fitting and Validation
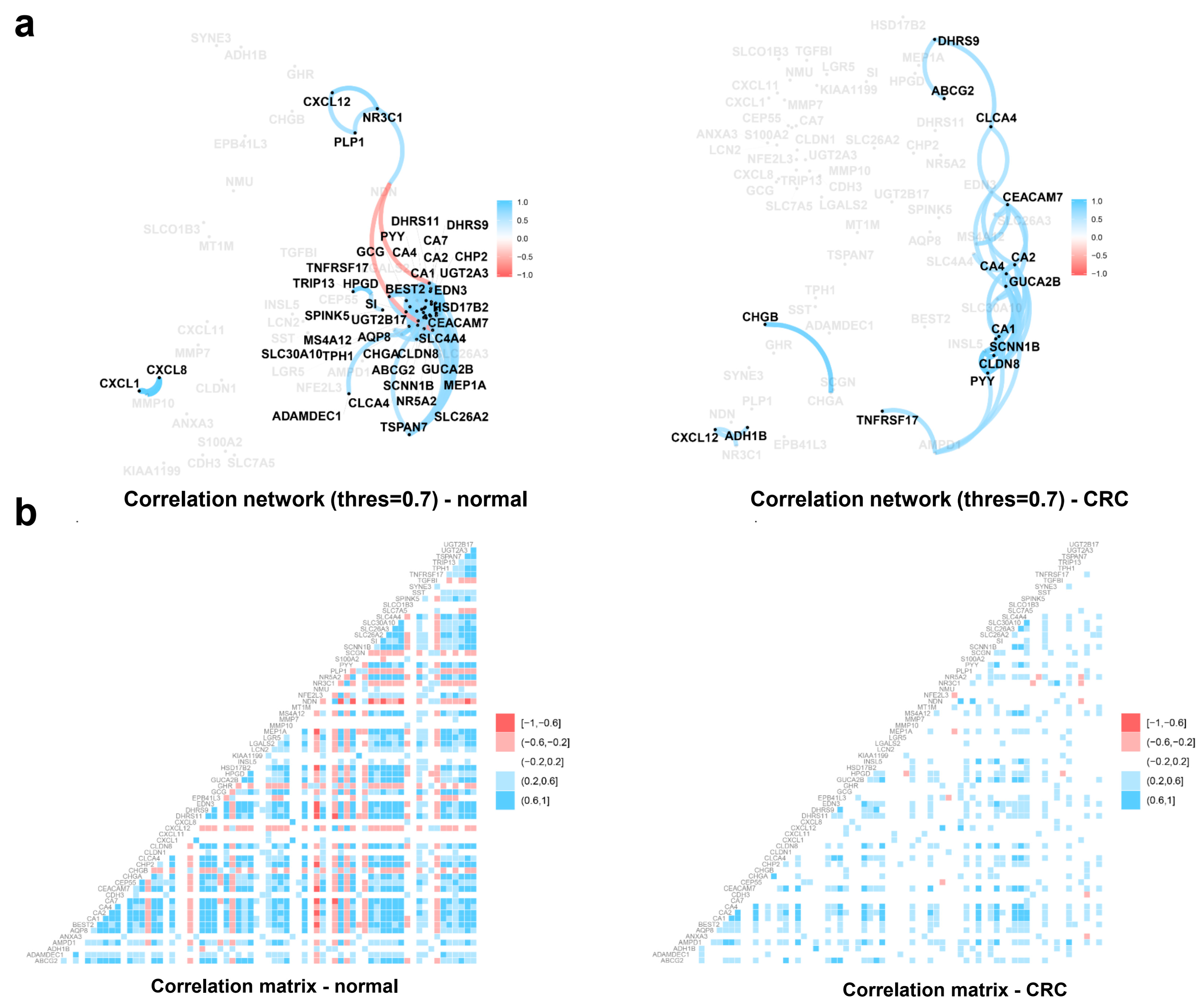
2.3. Functional Analysis of Individual Biomarkers in Colorectal Cancer
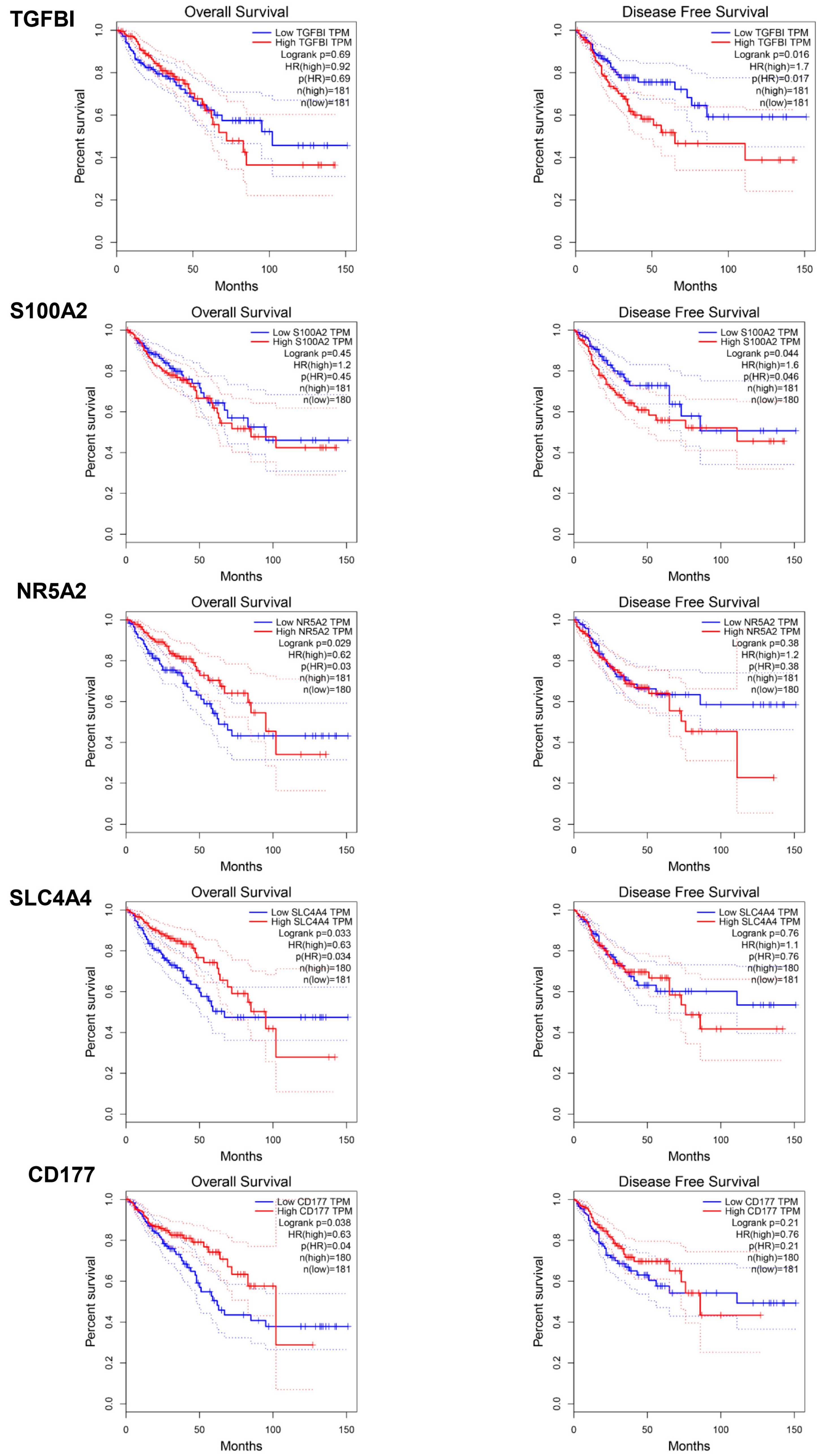
2.4. Prognostic Assessment of Individual Biomarkers in Colorectal Cancer
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Data Pre-Processing
4.3. Gene Expression Analysis
4.4. Data Exploration and Visualization
4.5. Variable Selection Method
4.6. Classification Model Fitting and Validation
4.7. Correlation Network Analysis
4.8. Survival Analysis
4.9. Functional Analysis
4.10. Statistical Significance Level
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CRC | Colorectal cancer |
| TCGA | The Cancer Genome Atlas |
| VIM | Variable importance measures |
| AUC | Area under the receiver operating characteristic curve |
| RF | Random forest |
| AUCRF | The area under the receiver operating characteristic curve of the random forest |
| GTEx | Genotype-Tissue Expression |
| PCA | Principal component analysis |
| CHAT | Cancer Hallmarks Analytics Tool |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GEO | Gene Expression Omnibus |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, M.; Gies, A.; Werner, S.; Schrotz-King, P.; Brenner, H. Blood-Based Protein Signatures for Early Detection of Colorectal Cancer: A Systematic Review. Clin. Transl. Gastroenterol. 2017, 8, e128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hibner, G.; Kimsa-Furdzik, M.; Francuz, T. Relevance of MicroRNAs as Potential Diagnostic and Prognostic Markers in Colorectal Cancer. Int. J. Mol. Sci. 2018, 19, 2944. [Google Scholar] [CrossRef] [PubMed]
- Rubin, G.; Walter, F.; Emery, J.; de Wit, N. Reimagining the diagnostic pathway for gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 181. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Han, R.; Han, Y.; Shi, X.; Hu, J.; Lu, B. Accuracy of Combined Computed Tomography Colonography and Dual Energy Iiodine Map Imaging for Detecting Colorectal masses using High-pitch Dual-source CT. Sci. Rep. 2018, 8, 3790. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Endo, S.; Honda, T.; Yasuda, T.; Hirayama, M.; Takahashi, S.; Kato, T.; Horita, S.; Furuya, K.; Kasai, K.; et al. Accuracy of CT Colonography for Detection of Polypoid and Nonpolypoid Neoplasia by Gastroenterologists and Radiologists: A Nationwide Multicenter Study in Japan. Am. J. Gastroenterol. 2016, 112, 163. [Google Scholar] [CrossRef]
- Thomsen, M.; Skovlund, E.; Sorbye, H.; Bolstad, N.; Nustad, K.J.; Glimelius, B.; Pfeiffer, P.; Kure, E.H.; Johansen, J.S.; Tveit, K.M.; et al. Prognostic role of carcinoembryonic antigen and carbohydrate antigen 19-9 in metastatic colorectal cancer: A BRAF-mutant subset with high CA 19-9 level and poor outcome. Br. J. Cancer 2018, 118, 1609–1616. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zhou, Y.; Sheng, S.; Qian, S.Y.; Huo, X. Evaluation of Serum CEA, CA19-9, CA72-4, CA125 and Ferritin as Diagnostic Markers and Factors of Clinical Parameters for Colorectal Cancer. Sci. Rep. 2018, 8, 2732. [Google Scholar] [CrossRef]
- Long, N.P.; Jung, K.H.; Yoon, S.J.; Anh, N.H.; Nghi, T.D.; Kang, Y.P.; Yan, H.H.; Min, J.E.; Hong, S.S.; Kwon, S.W. Systematic assessment of cervical cancer initiation and progression uncovers genetic panels for deep learning-based early diagnosis and proposes novel diagnostic and prognostic biomarkers. Oncotarget 2017, 8, 109436–109456. [Google Scholar] [CrossRef]
- Lee, D.-K.; Long, N.P.; Jung, J.; Kim, T.J.; Na, E.; Kang, Y.P.; Kwon, S.W.; Jang, J. Integrative lipidomic and transcriptomic analysis of X-linked adrenoleukodystrophy reveals distinct lipidome signatures between adrenomyeloneuropathy and childhood cerebral adrenoleukodystrophy. Biochem. Biophys. Res. Commun. 2018. [Google Scholar] [CrossRef] [PubMed]
- Collins, G.S.; Reitsma, J.B.; Altman, D.G.; Moons, K.M. Transparent reporting of a multivariable prediction model for individual prognosis or diagnosis (tripod): The tripod statement. Ann. Intern. Med. 2015, 162, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Novianti, P.W.; Jong, V.L.; Roes, K.C.B.; Eijkemans, M.J.C. Meta-analysis approach as a gene selection method in class prediction: Does it improve model performance? A case study in acute myeloid leukemia. BMC Bioinform. 2017, 18, 210. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, S.; Huang, C.C.; Yu, M.; Shao, J. Meta-analysis based variable selection for gene expression data. Biometrics 2014, 70, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Shabalin, A.A.; Tjelmeland, H.; Fan, C.; Perou, C.M.; Nobel, A.B. Merging two gene-expression studies via cross-platform normalization. Bioinformatics 2008, 24, 1154–1160. [Google Scholar] [CrossRef] [Green Version]
- Long, N.P.; Yoon, S.J.; Anh, N.H.; Nghi, T.D.; Lim, D.K.; Hong, Y.J.; Hong, S.-S.; Kwon, S.W. A systematic review on metabolomics-based diagnostic biomarker discovery and validation in pancreatic cancer. Metabolomics 2018, 14, 109. [Google Scholar] [CrossRef]
- Boulesteix, A.-L.; Janitza, S.; Kruppa, J.; König, I.R. Overview of random forest methodology and practical guidance with emphasis on computational biology and bioinformatics. Wiley Interdiscip. Rev. Data Min. Knowl. Discov. 2012, 2, 493–507. [Google Scholar] [CrossRef] [Green Version]
- Janitza, S.; Strobl, C.; Boulesteix, A.-L. An AUC-based permutation variable importance measure for random forests. BMC Bioinform. 2013, 14, 119. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Kursa, M.B.; Rudnicki, W.R. Feature Selection with the Boruta Package. J. Stat. Softw. 2010, 36, 13. [Google Scholar] [CrossRef]
- Janitza, S.; Celik, E.; Boulesteix, A.-L. A computationally fast variable importance test for random forests for high-dimensional data. Adv. Data Anal. Classif. 2016. [Google Scholar] [CrossRef]
- Ryan, B.M.; Zanetti, K.A.; Robles, A.I.; Schetter, A.J.; Goodman, J.; Hayes, R.B.; Huang, W.Y.; Gunter, M.J.; Yeager, M.; Burdette, L.; et al. Germline variation in NCF4, an innate immunity gene, is associated with an increased risk of colorectal cancer. Int. J. Cancer 2014, 134, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Sheffer, M.; Bacolod, M.D.; Zuk, O.; Giardina, S.F.; Pincas, H.; Barany, F.; Paty, P.B.; Gerald, W.L.; Notterman, D.A.; Domany, E. Association of survival and disease progression with chromosomal instability: A genomic exploration of colorectal cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 7131–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.; Park, M.; Jang, M.; Yun, S.; Kim, W.K.; Kim, S.; Paik, S.; Lee, H.J.; Hong, S.; Kim, T.I.; et al. Prognosis of stage III colorectal carcinomas with FOLFOX adjuvant chemotherapy can be predicted by molecular subtype. Oncotarget 2017, 8, 39367–39381. [Google Scholar] [CrossRef] [PubMed]
- Sabates-Bellver, J.; Van der Flier, L.G.; de Palo, M.; Cattaneo, E.; Maake, C.; Rehrauer, H.; Laczko, E.; Kurowski, M.A.; Bujnicki, J.M.; Menigatti, M.; et al. Transcriptome profile of human colorectal adenomas. Mol. Cancer Res. 2007, 5, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Armenia, J.; Zhang, C.; Penson, A.V.; Reznik, E.; Zhang, L.; Minet, T.; Ochoa, A.; Gross, B.E.; Iacobuzio-Donahue, C.A.; et al. Unifying cancer and normal RNA sequencing data from different sources. Sci. Data 2018, 5, 180061. [Google Scholar] [CrossRef] [PubMed]
- Bondurant, K.L.; Lundgreen, A.; Herrick, J.S.; Kadlubar, S.; Wolff, R.K.; Slattery, M.L. Interleukin genes and associations with colon and rectal cancer risk and overall survival. Int. J. Cancer 2013, 132, 905–915. [Google Scholar] [CrossRef]
- Rung, J.; Brazma, A. Reuse of public genome-wide gene expression data. Nat. Rev. Genet. 2012, 14, 89. [Google Scholar] [CrossRef]
- Walsh, C.J.; Hu, P.; Batt, J.; Santos, C.C. Microarray Meta-Analysis and Cross-Platform Normalization: Integrative Genomics for Robust Biomarker Discovery. Microarrays 2015, 4, 389–406. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Pena, M.L.; Isaza, C.E.; Perez-Morales, J.; Rodriguez-Padilla, C.; Castro, J.M.; Cabrera-Rios, M. Identification of potential biomarkers from microarray experiments using multiple criteria optimization. Cancer Med. 2013, 2, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Lin, C.W.; Tseng, G.C. MetaKTSP: A meta-analytic top scoring pair method for robust cross-study validation of omics prediction analysis. Bioinformatics 2016, 32, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Yao, C.T.; Su, S.L.; Chou, Y.C.; Chu, C.M.; Huang, C.S.; Terng, H.J.; Chou, H.L.; Wetter, T.; Chen, K.H.; et al. Verification of gene expression profiles for colorectal cancer using 12 internet public microarray datasets. World J. Gastroenterol. 2014, 20, 17476–17482. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bilbao, A.; Armananzas, R.; Ispizua, Z.; Calvo, B.; Alonso-Varona, A.; Inza, I.; Larranaga, P.; Lopez-Vivanco, G.; Suarez-Merino, B.; Betanzos, M. Identification of a biomarker panel for colorectal cancer diagnosis. BMC Cancer 2012, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Kapetanakis, N.; Kountouras, J.; Zavos, C.; Polyzos, S.A.; Kouklakis, G.; Venizelos, I.; Nikolaidou, C.; Vardaka, E.; Paikos, D.; Katsinelos, P. Helicobacter pylori infection and colorectal carcinoma: Pathologic aspects. J. Gastrointest. Oncol. 2012, 3, 377. [Google Scholar] [CrossRef] [PubMed]
- Kountouras, J.; Polyzos, S.A.; Doulberis, M.; Zeglinas, C.; Artemaki, F.; Vardaka, E.; Deretzi, G.; Giartza-Taxidou, E.; Tzivras, D.; Vlachaki, E.; et al. Potential impact of Helicobacter pylori-related metabolic syndrome on upper and lower gastrointestinal tract oncogenesis. Metabolism 2018, 87, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Camarda, R.; Odegaard, J.; Paik, H.; Oskotsky, B.; Krings, G.; Goga, A.; Sirota, M.; Butte, A.J. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat. Commun. 2017, 8, 1077. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.T.; Sameer, S.; Guestrin, C. “Why Should I Trust You?”: Explaining the Predictions of Any Classifier. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016. [Google Scholar]
- Xia, J.; Gill, E.E.; Hancock, R.E.W. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823. [Google Scholar] [CrossRef]
- Tang, Y.; Horikoshi, M.; Li, W. ggfortify: Unified Interface to Visualize Statistical Result of Popular R Packages. RJ 2016, 8, 474–485. [Google Scholar]
- Zhou, G.; Xia, J. OmicsNet: A web-based tool for creation and visual analysis of biological networks in 3D space. Nucleic Acids Res. 2018, 46, W514–W522. [Google Scholar] [CrossRef]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Degenhardt, F.; Seifert, S.; Szymczak, S. Evaluation of variable selection methods for random forests and omics data sets. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin, Germany, 2016. [Google Scholar]
- Jackson, S. corrr: Correlations in R. R Package Version 0.3.0. 2018. Available online: https://CRAN.R-project.org/package=corrr (accessed on 10 October 2018).
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.; Ali, I.; Silins, I.; Pyysalo, S.; Guo, Y.; Högberg, J.; Stenius, U.; Korhonen, A. Cancer Hallmarks Analytics Tool (CHAT): A text mining approach to organize and evaluate scientific literature on cancer. Bioinformatics 2017, 33, 3973–3981. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Section | Comparison | Author | Data Set | Year | Platform | Samples | ||
|---|---|---|---|---|---|---|---|---|
| Variable selection | Cancer versus Normalcy | Normalcy | Adenoma | Cancer | ||||
| Ryan BM et al. [22] | GSE44861 1 | 2013 | Affymetrix U133A | 55 | 56 | |||
| Sheffer M et al. [23] | GSE41258 1 | 2012 | Affymetrix U133A | 44 | 183 | |||
| Kwon Y et al. [24] | GSE83889 1 | 2016 | Illumina HumanHT-12 V4.0 | 35 | 101 | |||
| Adenoma versus Normalcy | Marra G et al. [25] | GSE8671 1 | 2007 | Affymetrix U133 2.0 | 32 | 32 | ||
| Model fitting and validation | Author | Type | Non-malignancy | Cancer | ||||
| Cancer versus Non-malignant | TCGA, GTEx [26] | coad-rsem-fpkm-tcga, coad-rsem-fpkm-tcga-t, read-rsem-fpkm-tcga, read-rsem-fpkm-tcga-t, colon-rsem-fpkm-gtex | 390 2 | 372 3 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, N.P.; Park, S.; Anh, N.H.; Nghi, T.D.; Yoon, S.J.; Park, J.H.; Lim, J.; Kwon, S.W. High-Throughput Omics and Statistical Learning Integration for the Discovery and Validation of Novel Diagnostic Signatures in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 296. https://doi.org/10.3390/ijms20020296
Long NP, Park S, Anh NH, Nghi TD, Yoon SJ, Park JH, Lim J, Kwon SW. High-Throughput Omics and Statistical Learning Integration for the Discovery and Validation of Novel Diagnostic Signatures in Colorectal Cancer. International Journal of Molecular Sciences. 2019; 20(2):296. https://doi.org/10.3390/ijms20020296
Chicago/Turabian StyleLong, Nguyen Phuoc, Seongoh Park, Nguyen Hoang Anh, Tran Diem Nghi, Sang Jun Yoon, Jeong Hill Park, Johan Lim, and Sung Won Kwon. 2019. "High-Throughput Omics and Statistical Learning Integration for the Discovery and Validation of Novel Diagnostic Signatures in Colorectal Cancer" International Journal of Molecular Sciences 20, no. 2: 296. https://doi.org/10.3390/ijms20020296
APA StyleLong, N. P., Park, S., Anh, N. H., Nghi, T. D., Yoon, S. J., Park, J. H., Lim, J., & Kwon, S. W. (2019). High-Throughput Omics and Statistical Learning Integration for the Discovery and Validation of Novel Diagnostic Signatures in Colorectal Cancer. International Journal of Molecular Sciences, 20(2), 296. https://doi.org/10.3390/ijms20020296