Activity to Breast Cancer Cell Lines of Different Malignancy and Predicted Interaction with Protein Kinase C Isoforms of Royleanones

 ,
,  ,
,  ,
,  , ,
, ,  and
and 
Abstract
:
1. Introduction
2. Results
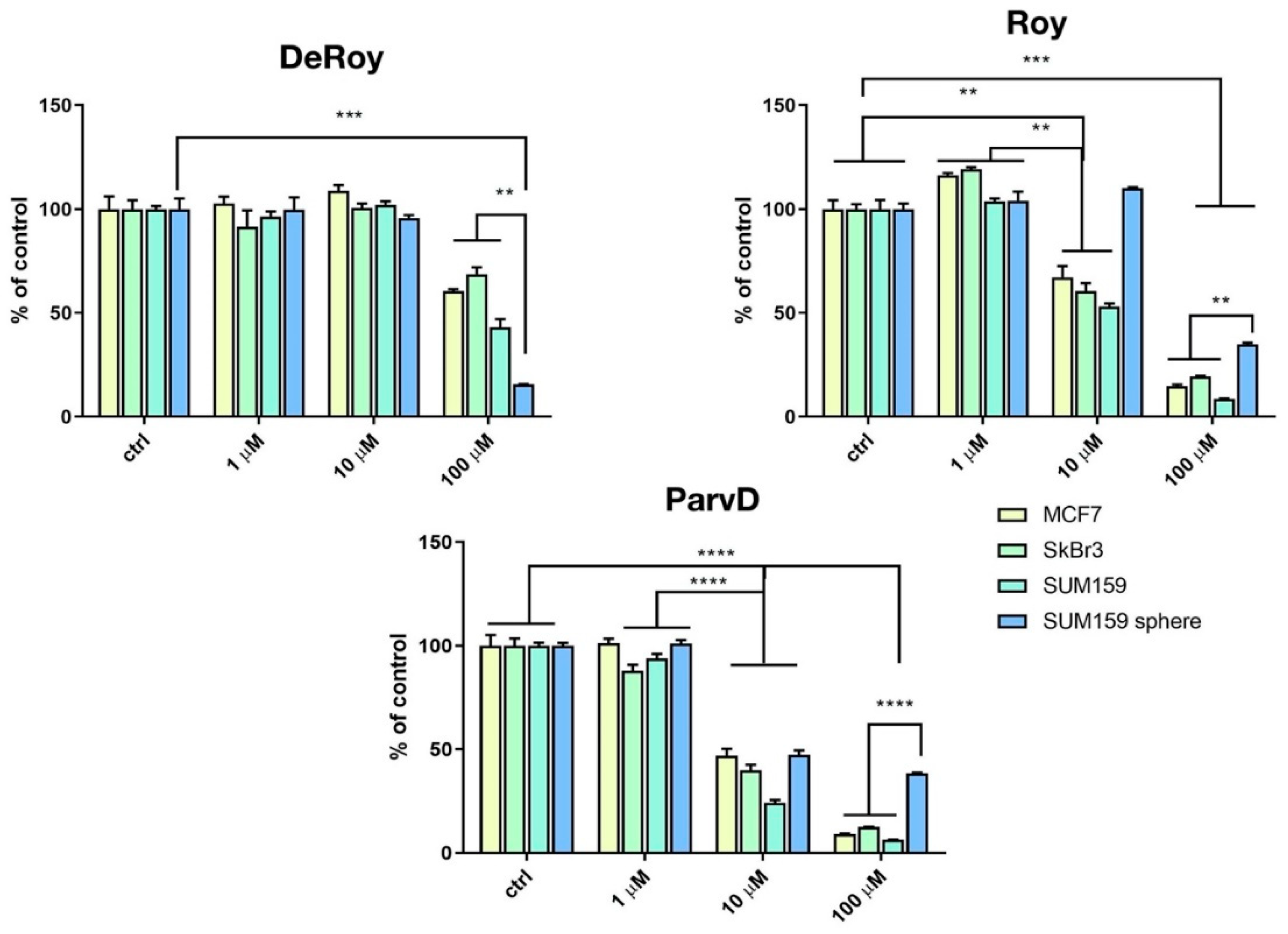
2.1. MTT Breast Cancer
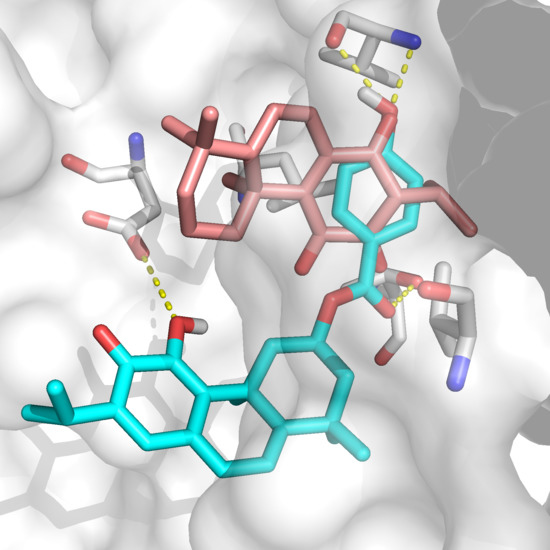
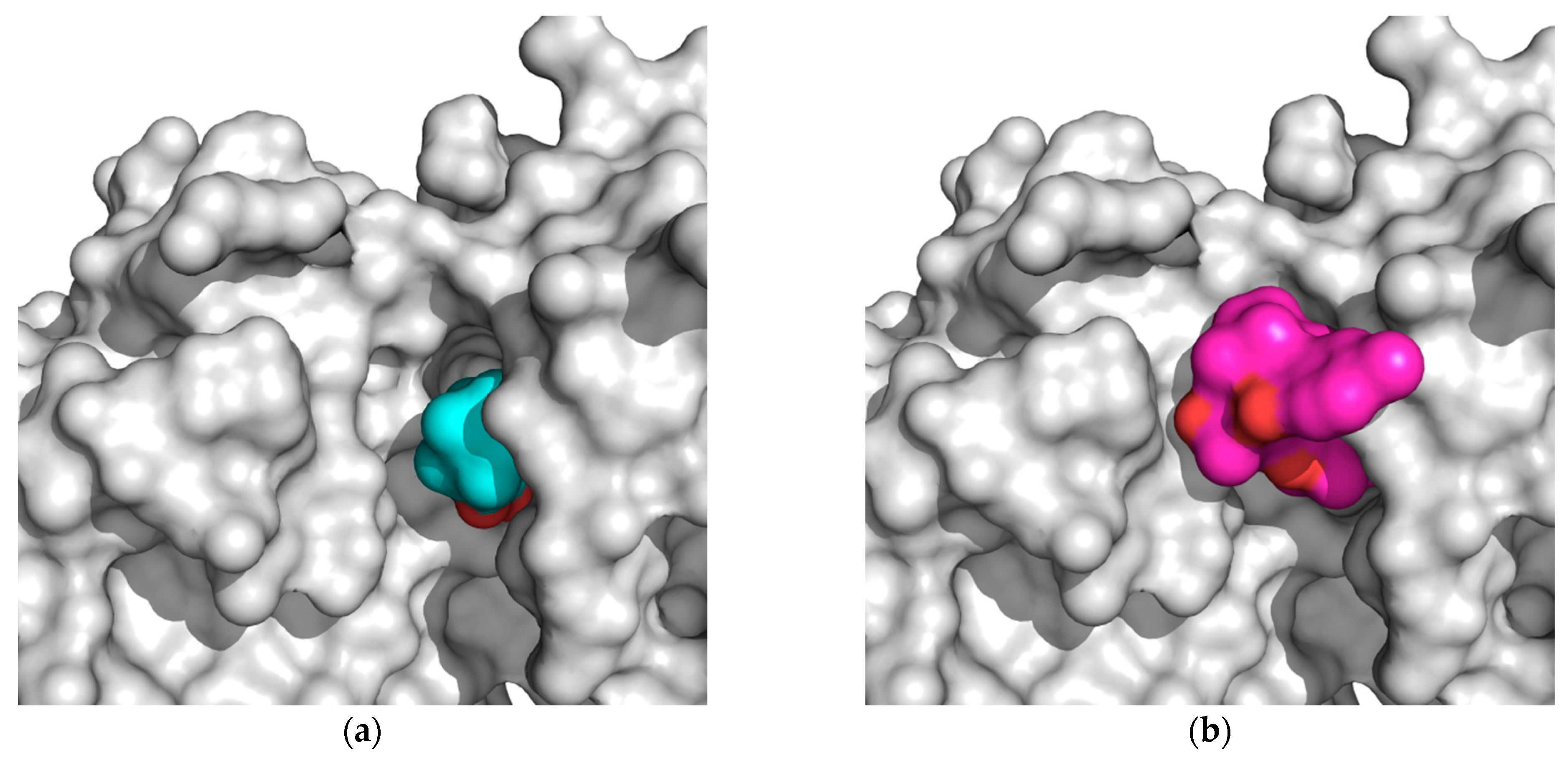
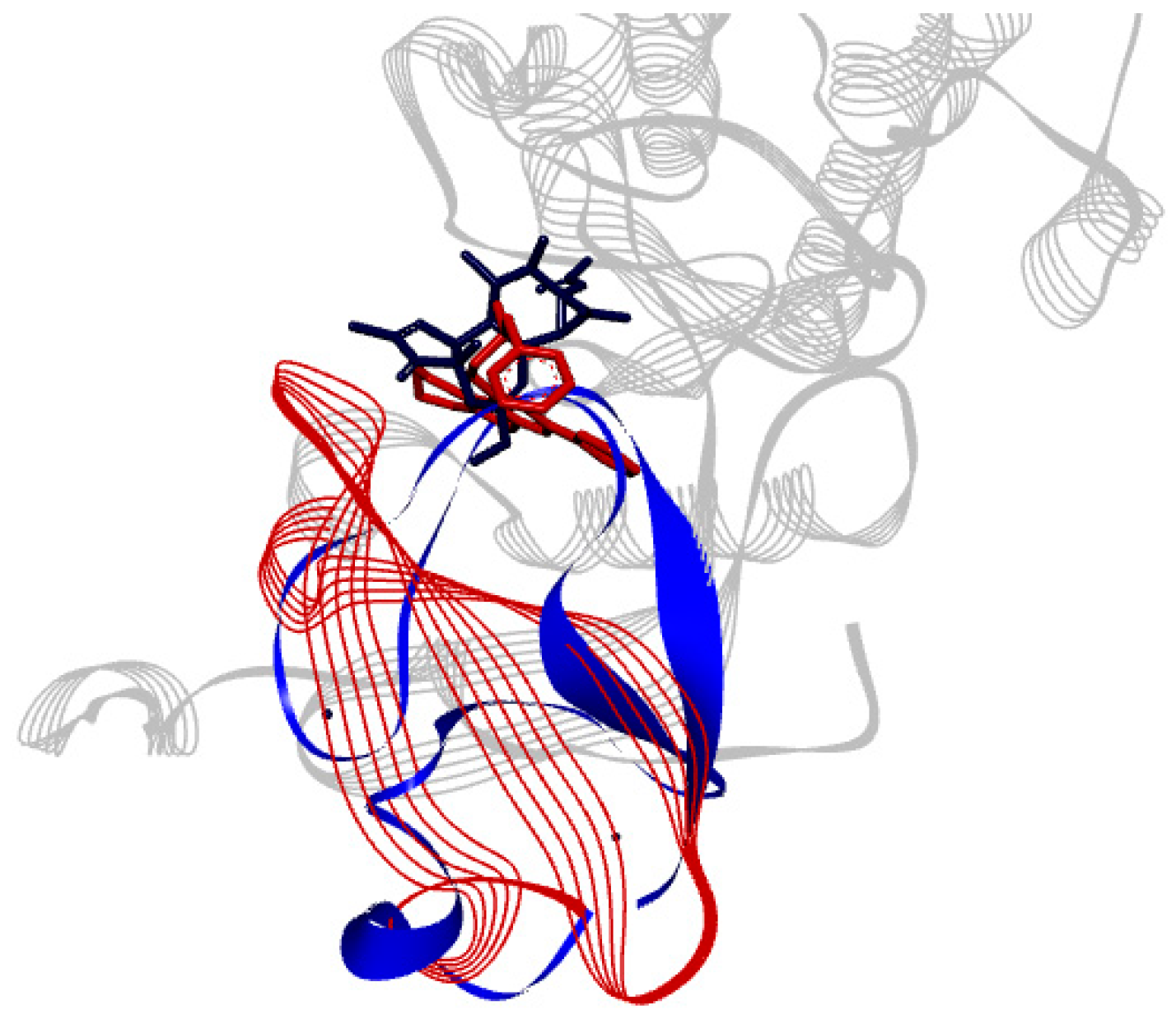
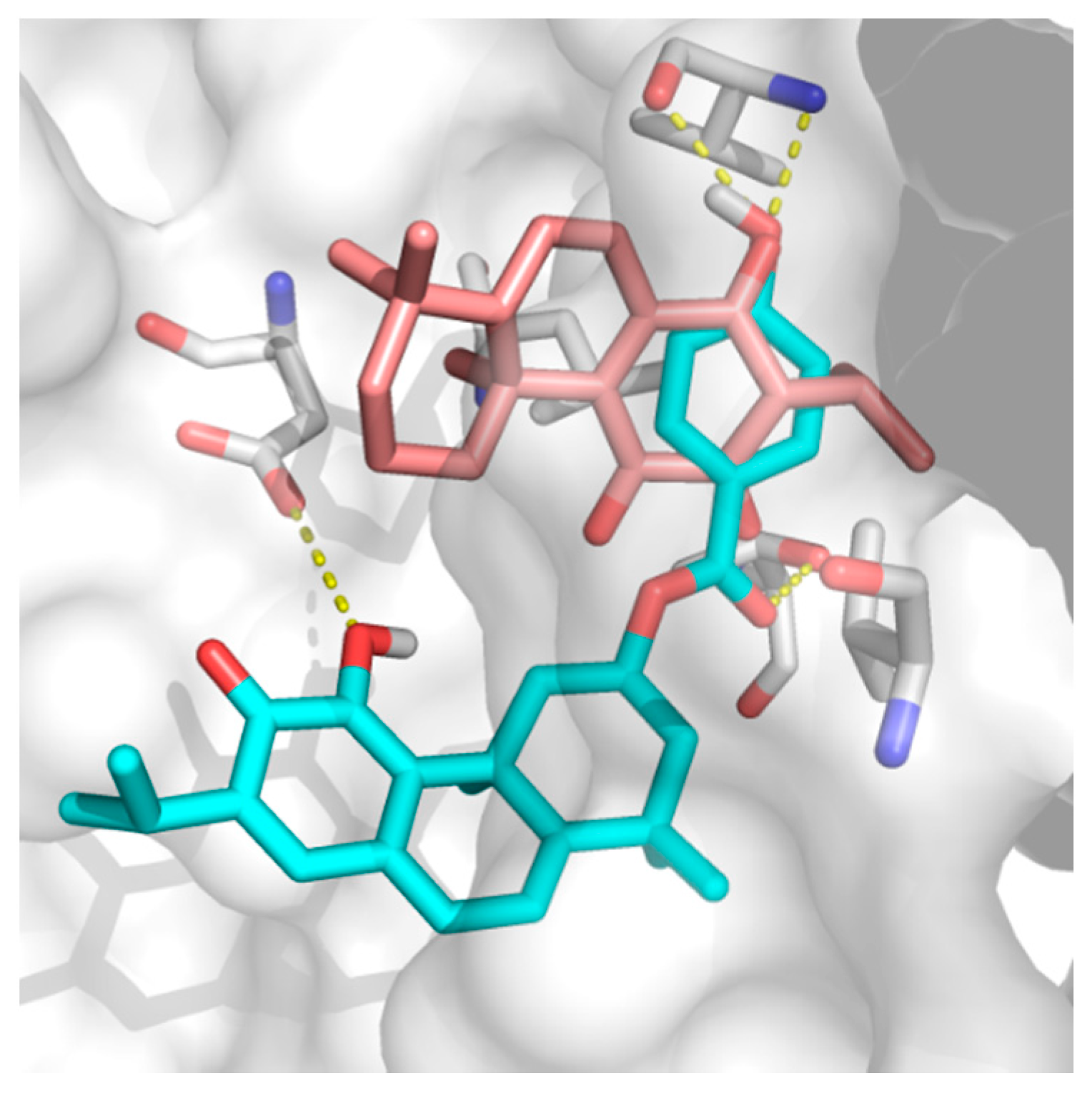
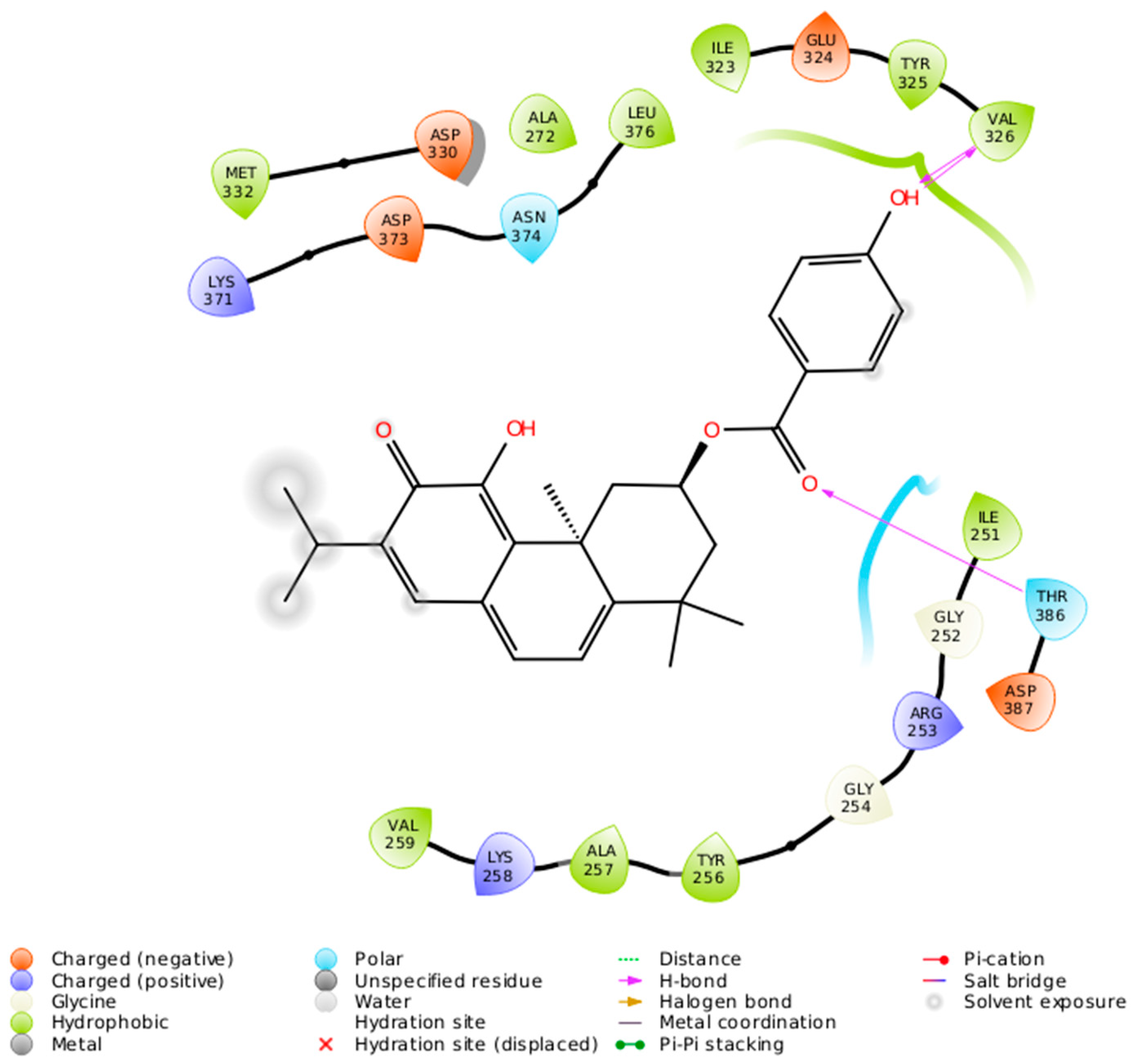
2.2. Docking Results
3. Discussion
4. Materials and Methods
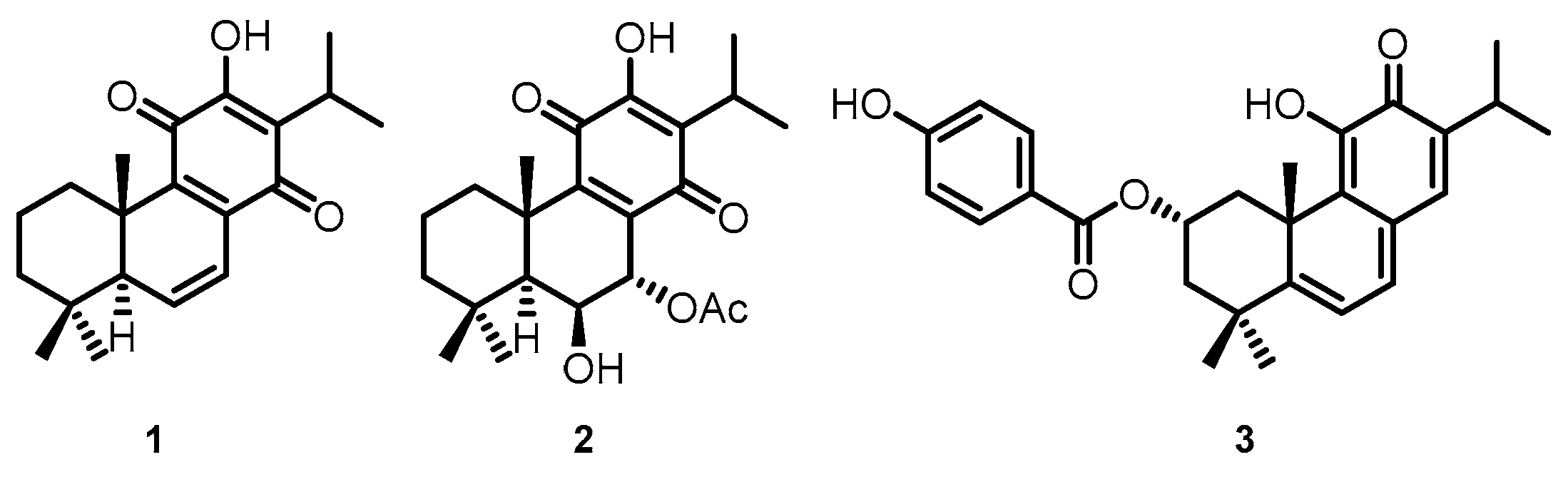
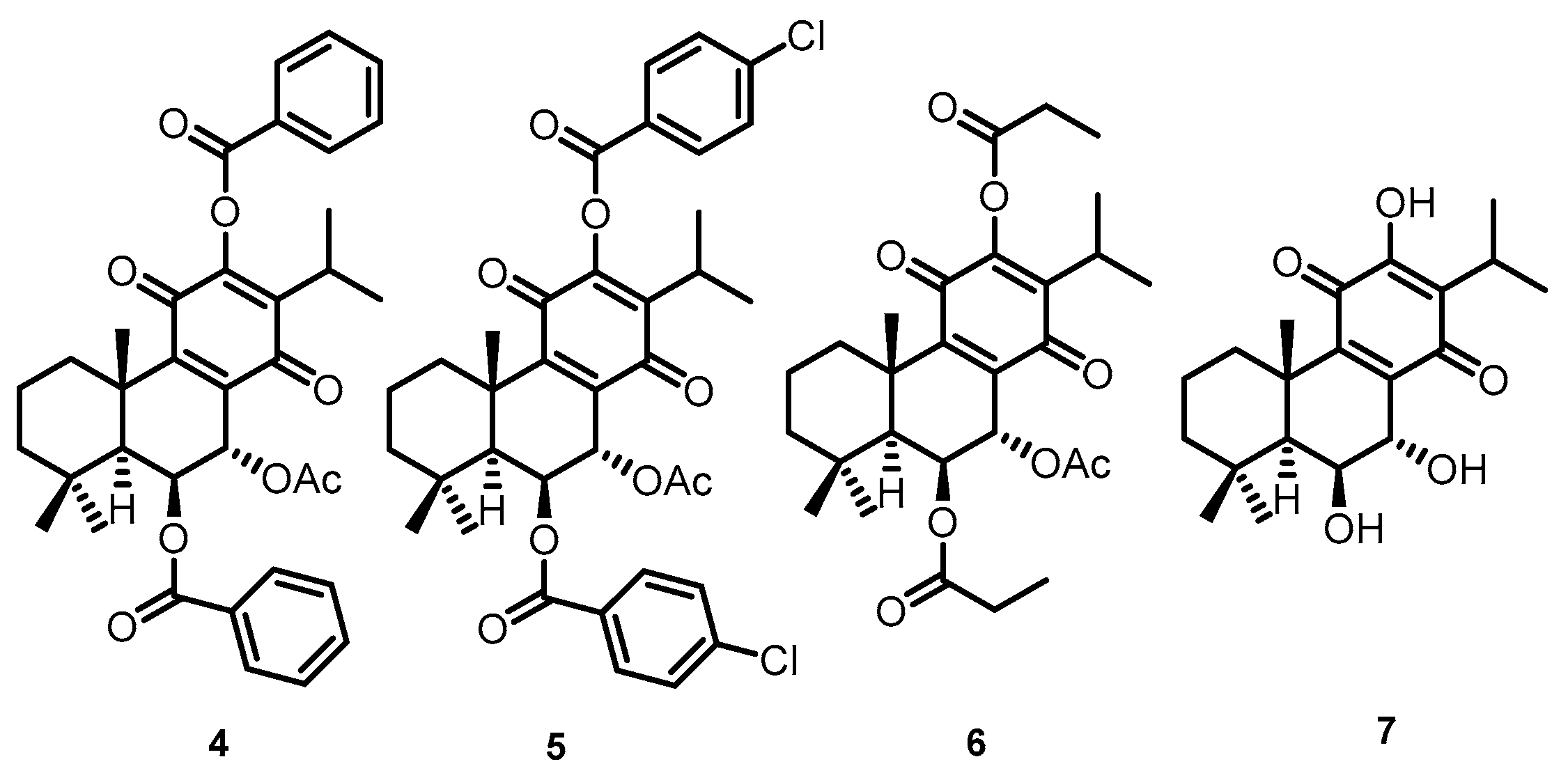
4.1. Compounds
4.2. Cytotoxicity Assay
4.3. Cell Viability Assay
4.4. Ligands
4.5. Sequence Alignment
4.6. Docking
4.7. Vina
4.8. Glide
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Demain, A.L.; Vaishnav, P. Natural Products for Cancer Chemotherapy. Microb. Biotechnol. 2011, 4, 687–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajesh, E.; Sankari, L.S.; Malathi, L.; Krupaa, J.R. Naturally Occurring Products in Cancer Therapy. J. Pharm. Bioallied Sci. 2015, 7, S181–S183. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.; Silva, C.O.; Monteiro, C.M.; Nicolai, M.; Viana, A.; Andrade, J.M.; Barasoain, I.; Stankovic, T.; Quintana, J.; Hernández, I.; et al. Anticancer Properties of the Abietane Diterpene 6,7-Dehydroroyleanone Obtained by Optimized Extraction. Future Med. Chem. 2018, 1, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Lukhoba, C.W.; Simmonds, M.S.J.; Paton, A.J. Plectranthus: A Review of Ethnobotanical Uses. J. Ethnopharmacol. 2006, 103, 1–24. [Google Scholar] [CrossRef]
- Rice, L.J.; Brits, G.J.; Potgieter, C.J.; Van Staden, J. Plectranthus: A Plant for the Future? S. Afr. J. Bot. 2011, 77, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Ladeiras, D.; Monteiro, C.M.; Pereira, F.; Reis, C.P.; Afonso, C.A.M.; Rijo, P. Reactivity of Diterpenoid Quinones: Royleanones. Curr. Pharm. Des. 2016, 22, 1682–1714. [Google Scholar] [CrossRef]
- Matias, D.; Nicolai, M.; Costa, J.; Saraiva, N.; Fernandes, A.S.; Simões, M.F.; Diaz-Lanza, A.M.; Reis, C.P.; Rijo, P. Cytotoxicity Screening of Plectranthus spp. Extracts and Individual Components in MDA-MB-231 Cells. Toxicol. Lett. 2015, 238, S240. [Google Scholar] [CrossRef]
- Rijo, P.; Fernandes, A.S.; Simões, F.; Pinheiro, L. Evaluation of Diterpenoids from P. Ornatus as Potential COX-1 Inhibitors. J. Investig. Biomédica e Biofarm. 2012, 9, 111–118. [Google Scholar] [CrossRef]
- Marques, C.G.; Pedro, M.; Simões, M.F.; Nascimento, M.S.; Pinto, M.M.; Rodríguez, B. Effect of Abietane Diterpenes from Plectranthus grandidentatus on the Growth of Human Cancer Cell Lines. Planta Med. 2002, 68, 839–840. [Google Scholar] [CrossRef]
- Burmistrova, O.; Perdomo, J.; Simões, M.F.; Rijo, P.; Quintana, J.; Estévez, F. The Abietane Diterpenoid Parvifloron D from Plectranthus Ecklonii is a Potent Apoptotic Inducer in Human Leukemia Cells. Phytomedicine 2015, 22, 1009–1016. [Google Scholar] [CrossRef]
- Rijo, P.; Duarte, A.; Francisco, A.P.; Semedo-Lemsaddek, T.; Simões, M.F. In Vitro Antimicrobial Activity of Royleanone Derivatives Against Gram-Positive Bacterial Pathogens. Phyther. Res. 2014, 28, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Kubínová, R.; Pořízková, R.; Navrátilová, A.; Farsa, O.; Hanáková, Z.; Bačinská, A.; Čížek, A.; Valentová, M.; Cížek, A.; Valentová, M.; et al. Antimicrobial and Enzyme Inhibitory Activities of the Constituents of Plectranthus madagascariensis (Pers.) Benth. J. Enzyme Inhib. Med. Chem. 2014, 6366, 1–4. [Google Scholar] [CrossRef]
- Gazim, Z.C.; Rodrigues, F.; Amorin, A.C.L.; De Rezende, C.M.; Sokovic, M.; Teševic, V.; Vuckovic, I.; Krstic, G.; Cortez, L.E.R.; Colauto, N.B.; et al. New Natural Diterpene-Type Abietane from Tetradenia Riparia Essential Oil with Cytotoxic and Antioxidant Activities. Molecules 2014, 19, 514–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burmistrova, O.; Simões, M.F.; Rijo, P.; Quintana, J.; Bermejo, J.; Estévez, F. Antiproliferative Activity of Abietane Diterpenoids against Human Tumor Cells. J. Nat. Prod. 2013, 76, 1413–1423. [Google Scholar] [CrossRef] [Green Version]
- Bessa, C.; Soares, J.; Raimundo, L.; Loureiro, J.B.; Gomes, C.; Reis, F.; Soares, M.L.; Santos, D.; Dureja, C.; Chaudhuri, S.R.; et al. Discovery of a Small-Molecule Protein Kinase Cδ-Selective Activator with Promising Application in Colon Cancer Therapy Article. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Saraiva, L.; Rijo, P. Roy-Bz: A Small Molecule Activator of Protein Kinase C Delta. PCT/IB2017/050633, 10 May 2017. [Google Scholar]
- Matias, D.; Bessa, C.; Fátima Simões, M.; Reis, C.P.; Saraiva, L.; Rijo, P. Chapter 2—Natural Products as Lead Protein Kinase C Modulators for Cancer Therapy. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elseiver: Karachi, Pakistan, 2016; Volume 50, pp. 45–79. [Google Scholar] [CrossRef]
- Marengo, B.; De Ciucis, C.; Ricciarelli, R.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Protein Kinase C: An Attractive Target for Cancer Therapy. Cancers (Basel) 2011, 3, 531–567. [Google Scholar] [CrossRef] [Green Version]
- Griner, E.M.; Kazanietz, M.G. Protein Kinase C and Other Diacylglycerol Effectors in Cancer. Nat. Rev. Cancer 2007, 7, 281–294. [Google Scholar] [CrossRef]
- Cooke, M.; Magimaidas, A.; Casado-Medrano, V.; Kazanietz, M.G. Protein Kinase C in Cancer: The Top Five Unanswered Questions. Mol. Carcinog. 2017, 56, 1531–1542. [Google Scholar] [CrossRef]
- Garg, R.; Benedetti, L.G.; Abera, M.B.; Wang, H.; Abba, M.; Kazanietz, M.G. Protein Kinase C and Cancer: What We Know and What We Do Not. Oncogene 2014, 33, 5225–5237. [Google Scholar] [CrossRef] [Green Version]
- Mandil, R.; Ashkenazi, E.; Blass, M.; Kronfeld, I.; Kazimirsky, G.; Rosenthal, G.; Umansky, F.; Lorenzo, P.S.; Blumberg, P.M.; Brodie, C. Protein Kinase Cα and Protein Kinase Cδ Play Opposite Roles in the Proliferation and Apoptosis of Glioma Cells. Cancer Res. 2001, 61, 4612–4619. [Google Scholar] [PubMed]
- Teicher, B.; Menon, K.; Alvarez, E.; Shih, C.; Faul, M. Antiangiogenic and Antitumor Effects of a Protein Kinase Cβ Inhibitor in Human Breast Cancer and Ovarian Cancer Xenografts. Invest. New Drugs 2002, 20, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Murray, N.; Weems, C.; Chen, L.; Guo, H.; Ethridge, R.; Ceci, J.; Evers, B.; Thompson, E.; Fields, A. Role of Cyclooxygenase 2 in Protein Kinase C II-Mediated Colon Carcinogenesis. J. Biol. Chem. 2003, 278, 11167–11174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Choi, Y.-L.; Vallentin, A.; Hunrichs, B.; Hellerstein, M.; Peehl, D.; Mochly-Rosen, D. Centrosomal PKC II and Pericentrin Are Critical for Human Prostate Cancer Growth and Angiogenesis. Cancer Res. 2008, 68, 6831–6839. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-H.; Chung, C.; Kim, J.-M.; Lee, D.; Cho, S.Y.; Lee, T.H.; Cho, H.J.; Yeo, M.-K. Clinical Significance of Atypical Protein Kinase C (PKCι and PKCζ) and Its Relationship with Yes-Associated Protein in Lung Adenocarcinoma. BMC Cancer 2019, 19, 804. [Google Scholar] [CrossRef] [Green Version]
- Nanos-Webb, A.; Bui, T.; Karakas, C.; Zhang, D.; Carey, J.P.W.; Mills, G.B.; Hunt, K.K.; Keyomarsi, K. PKCiota Promotes Ovarian Tumor Progression through Deregulation of Cyclin E. Oncogene 2016, 35, 2428–2440. [Google Scholar] [CrossRef] [Green Version]
- Datta, J.; Smith, A.; Lang, J.C.; Islam, M.; Dutt, D.; Teknos, T.N.; Pan, Q. MicroRNA-107 Functions as a Candidate Tumor-Suppressor Gene in Head and Neck Squamous Cell Carcinoma by Downregulation of Protein Kinase Cɛ. Oncogene 2012, 31, 4045–4053. [Google Scholar] [CrossRef] [Green Version]
- Cooke, M.; Baker, M.; Kazanietz, M.; Casado-Medrano, V. PKCε Regulates Rho GTPases and Actin Cytoskeleton Reorganization in Non-Small Cell Lung Cancer Cells. Small GTPases 2019. [Google Scholar] [CrossRef]
- Paul, A.; Danley, M.; Saha, B.; Tawfik, O.; Paul, S. PKCζ Promotes Breast Cancer Invasion by Regulating Expression of E-Cadherin and Zonula Occludens-1 (ZO-1) via NFκB-P65. Sci. Rep. 2015, 5, 12520. [Google Scholar] [CrossRef] [Green Version]
- Storz, P. Targeting Protein Kinase C Subtypes in Pancreatic Cancer. Expert Rev. Anticancer Ther. 2015, 15, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Li, L.; Zhang, Y.; Yang, J.; Li, M.; Zeng, F.; Deng, F. PKCζ in Prostate Cancer Cells Represses the Recruitment and M2 Polarization of Macrophages in the Prostate Cancer Microenvironment. Tumor Biol. 2017, 39, 101042831770144. [Google Scholar] [CrossRef] [Green Version]
- Kinehara, M.; Kawamura, S.; Tateyama, D.; Suga, M.; Matsumura, H.; Mimura, S.; Hirayama, N.; Hirata, M.; Uchio-Yamada, K.; Kohara, A.; et al. Protein Kinase C Regulates Human Pluripotent Stem Cell Self-Renewal. PLoS ONE 2013, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Tam, W.L.; Lu, H.; Buikhuisen, J.; Soh, B.S.; Lim, E.; Reinhardt, F.; Wu, Z.J.; Krall, J.A.; Bierie, B.; Guo, W.; et al. Protein Kinase C α Is a Central Signaling Node and Therapeutic Target for Breast Cancer Stem Cells. Cancer Cell 2013, 24, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Wu-Zhang, A.X.; Newton, A.C. Protein Kinase C Pharmacology: Refining the Toolbox. Biochem. J. 2013, 452, 195–209. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.S.; Ali, S.; El-Rayes, B.F.; Philip, P.A.; Sarkar, F.H. Exploitation of Protein Kinase C: A Useful Target for Cancer Therapy. Cancer Treat. Rev. 2009, 35, 1–8. [Google Scholar] [CrossRef]
- Protein Data Bank. Research Collaboratory for Structural Bioinformatics. Available online: http://www.pdb.org/pdb/home/home.do (accessed on 16 December 2019).
- Kjaer, S.; Linch, M.; Purkiss, A.; Kostelecky, B.; Knowles, P.P.; Rosse, C.; Riou, P.; Soudy, C.; Kaye, S.; Patel, B.; et al. Adenosine-binding motif mimicry and cellular effects of a thieno[2,3-d]pyrimidine-based chemical inhibitor of atypical protein kinase C isoenzymes. Biochem. J. 2013, 451, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, S.F. Structural Basis of Protein Kinase C Isoform Function. Physiol Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef] [Green Version]
- Bernardes, C.E.S.; Garcia, C.; Pereira, F.; Mota, J.; Pereira, P.; Cebola, M.J.; Reis, C.P.; Correia, I.; Piedade, F.M.; da Piedade, M.E.M.; et al. Extraction Optimization, Structural and Thermal Characterization of the Antimicrobial Abietane 7α-Acetoxy-6β-Hydroxyroyleanone. Mol. Pharm. 2018, 5, 1412–1419. [Google Scholar] [CrossRef]
- Simões, M.F.; Rijo, P.; Duarte, A.; Matias, D.; Rodríguez, B. An Easy and Stereoselective Rearrangement of an Abietane Diterpenoid into a Bioactive Microstegiol Derivative. Phytochem. Lett. 2010, 3, 234–237. [Google Scholar] [CrossRef]
- Rijo, P.; Simões, M.F.; Francisco, A.P.; Rojas, R.; Gilman, R.H.; Vaisberg, A.J.; Rodríguez, B.; Moiteiro, C. Antimycobacterial Metabolites from Plectranthus: Royleanone Derivatives against Mycobacterium tuberculosis Strains. Chem. Biodivers. 2010, 7, 922–932. [Google Scholar] [CrossRef]
- Graph Prism 7.0 Software; GraphPad Software: La Jolla, CA, USA, 2019.
- PerkinElmer Inc. ChemDraw 17.0; PerkinElmer Inc.: Waltham, MA, USA, 2017. [Google Scholar]
- Schrödinger Software. Schrödinger Release 2019-4: MacroModel; Schrödinger Software: New York, NY, USA, 2019. [Google Scholar]
- AutoDock Vina. The Scripps Research Institute. Available online: http://vina.scripps.edu/ (accessed on 1 January 2020).
- AutoDock. The Scripps Research Institute. Available online: http://autodock.scripps.edu/resources/adt (accessed on 5 January 2020).
- UniProt Consortium. UniProt Database. Available online: https://www.uniprot.org/ (accessed on 7 January 2020).
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Software. Schrödinger Release 2018-2: Virtual Screening Workflow; Schrödinger Software: New York, NY, USA, 2018. [Google Scholar]
- García-Sosa, A.T.; Sild, S.; Takkis, K.; Maran, U. Combined Approach Using Ligand Efficiency, Cross-Docking, and Antitarget Hits for Wild-Type and Drug-Resistant Y181C HIV-1 Reverse Transcriptase. J. Chem. Inf. Model. 2011, 51, 2595–2611. [Google Scholar] [CrossRef]
- Viira, B.; Selyutina, A.; García-Sosa, A.T.; Karonen, M.; Sinkkonen, J.; Merits, A.; Maran, U. Design, Discovery, Modelling, Synthesis, and Biological Evaluation of Novel and Small, Low Toxicity s-Triazine Derivatives as HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors. Bioorg. Med. Chem. 2016, 24, 2519–2529. [Google Scholar] [CrossRef]
- García-Sosa, A.T.; Sild, S.; Maran, U. Docking and Virtual Screening Using Distributed Grid Technology. SQER 2009, 28, 815–821. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uniprot, Isoform | ζ | Ι | δ | θ | ε | α | β |
|---|---|---|---|---|---|---|---|
| sp|P41743|KPCI_HUMAN, ζ | 100 | 72.81 | 36.82 | 36.86 | 44.78 | 43.57 | 43.48 |
| sp|P41743|KPCI_HUMAN, ι | 72.81 | 100 | 36.14 | 36.52 | 43.91 | 45.36 | 44.21 |
| sp|Q05655|KPCD_HUMAN, δ | 36.82 | 36.14 | 100 | 64.89 | 43.88 | 47.61 | 48.99 |
| sp|Q04759|KPCT_HUMAN, θ | 36.86 | 36.52 | 64.89 | 100 | 43.31 | 48.51 | 47.64 |
| sp|Q02156|KPCE_HUMAN, ε | 44.78 | 43.91 | 43.88 | 43.31 | 100 | 52.9 | 53.14 |
| sp|P17252|KPCA_HUMAN, α | 43.57 | 45.36 | 47.61 | 48.51 | 52.9 | 100 | 79.01 |
| sp|P05771|KPCB_HUMAN, β | 43.48 | 44.21 | 48.99 | 47.64 | 53.14 | 79.01 | 100 |
| Compound | 5f9e (Isoform θ) | 2i0e (Isoform βII) | 3zh8 (Isoform ι) | 4ra4 (Isoform α) | 1ptr (Isoform δ) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vina | Glide XP | MMGBSA | Vina | Glide XP | MMGBSA | Vina | Glide XP | MMGBSA | Vina | Glide XP | MMGBSA | Vina | Glide XP | MMGBSA | |
| PMA | −7.4 | −8.0 | −4.84 | −40.13 | −7.2 | −6.4 | −4.7 | −4.41 | −42.12 | ||||||
| ARA | −6.3 | −7.8 | −1.00 | −40.95 | −6.5 | −5.6 | −4.4 | −1.85 | −18.56 | ||||||
| 5VS1001 (5f9e co-cryst.) | −10.5 | −7.2 | −40.48 | ||||||||||||
| PDS 902 (2i0e co-cryst.) | –11.0 | −10.02 | −56.23 | ||||||||||||
| C581582 (3zh8 co-cryst.) | −9.9 | −8.0 | −58.56 | ||||||||||||
| 3KZ701 (4ra4 co-cryst.) | −10.4 | −10.0 | 0 | ||||||||||||
| PRB3 (1ptr co-cryst.) | −6.3 | −4.25 | −27.00 | ||||||||||||
| 1 (DeRoy) | –9.3 | −5.8 | −44.29 | −12.0 | −6.21 | −30.81 | −8.4 | −5.6 | −36.23 | −8.4 | −6.7 | 0 | −6.2 | −4.13 | −29.39 |
| 2 (Roy) | –9 | −10.4 | −8.8 | −8 | −6.7 | ||||||||||
| 3 (ParvD) | –9.8 | −2.0 | −12.0 | −5.84 | −9.8 | −6.91 | −9.3 | −4.84 | −8.4 | −4.68 | |||||
| 4 (RoyBz) | –9.3 | −9.4 | −9.0 | −8.7 | −6.9 | ||||||||||
| 5 (RoyBzCl) | –8.8 | −9.8 | −9.4 | −8.4 | −6.7 | ||||||||||
| 6 (RoyPr2) | –8.7 | −7.9 | −7.4 | −7.5 | −6.3 | ||||||||||
| 7 (DihidroxyRoy) | –8.3 | −10.5 | −8.7 | −7.6 | −7.9 | ||||||||||
| Compounds | PKC Isoform | |||
|---|---|---|---|---|
| α | βI | ι | θ | |
| DeRoy (1) | Met 417 (L), Ala 480 (L), Met 470 (L), Lys 368 (H), Val 353 (L), Leu 345 (L) | Met 473 (L), Ala 483 (L), Phe 485 (L), Leu 394 (L), Phe 353 (L), Val 356 (L), Lys 371 (L), Phe 418 (L), Met 420 (L), Ala 369 (L), Leu 348 (L) | Leu 376 (L), Thr 386 (R), Val 307 (L), Phe 297 (L), Ile 323 (L), Lys 274 (L), Val 259 (L), Ile 251 (L) | Leu 511 (L), Ala 521 (L), Met 458 (L), Lys 409 (H), Val 394 (H), Phe 391 (R) |
| Roy (2) | Val 420 (H), Lys 368 (L), Ala 366 (L), Met 417 (L), Val 353 (L), Leu 345 (H, L) | Met 473 (L), Ala 483 (L), Phe 353 (L), Met 420 (L), Lys 371 (L), Val 356 (L) | Asp 373 (H), Ile 323 (L) | Leu 511 (L), Asn 509 (L), Ala 521 (L), Met 458 (L), Lys 409 (L), Val 394 (L), Leu 386 (L) |
| ParvD (3) | Met 470 (L), Val 353 (L), Ala 366 (L), Leu 345 (L), Met 417 (R, L), Lys 368 (L), Leu 391 (L), Ala 480 (L) | Met 473 (L), Tyr 422 (L), Leu 348 (L), Val 356 (L), Ala 483 (L), Ala 369 (L), Asn 471 (H), Phe 485 (L), Leu 394 (L), Lys 371 (L) | Ile 251 (L), Val 259 (L), Leu 376 (L), Tyr 325 (H), Val 259 (L), Thr 386 (L), Lys 274 (L), Ile 323 (L), Val 307 (L), Phe 297 (L) | Leu 511 (L), Ala 521 (L), Met 458 (L), Ala 407 (L), Val 394 (L), Phe 391 (L) |
| RoyBz (4) | Asp 424 (H), Ala 366 (L), Val 353 (L), Met 417 (L), Lys 368 (L), Ala 480 (L) | Leu 348 (L), Met 473 (L), Val 356 (L), Phe 353 (L), Lys 371 (L), Met 420 (L), Ala 483 (L) | Phe 333 (L, R), Asp 330 (H), Ile 251 (L), Leu 376 (L), Val 259 (L), Thr 386 (R), Ala 272 (L), Ile 323 (L), Val 307 (L) | Gly 464 (L), Phe 391 (L), Val 394 (L), Ala 407 (L), Met 458 (L), Ala 521 (L), Asp 522 (L), Lys 409 (H) |
| RoyBzCl (5) | Asp 424 (H), Gly 423 (L), Met 343 (R, L), Val 353 (L), Phe 350 (L), Lys 368 (L), Met 417 (L), Ala 480 (L) | Met 473 (L), Ala (483), Leu 394 (L), Met 420 (R, L), Lys 371 (L), Val 356 (L), Phe 353 (L), Leu 348 (L) | Phe 333 (R), Asp 330 (H), Thr 386 (R), Val 307 (L), Ile 323 (R), Ala 272 (R), Val 259 (R, L), Ile 251 (L), Arg 253 (R, L) | Leu 511 (L), Ala 521 (L), Lys 506 (L), Phe 391 (R), Val 394 (L), Leu 386 (R), Tyr 460 (L) |
| RoyPr2 (6) | Asp 424 (H), Met 470 (L), Val 420 (L), Met 417 (L), Ala 366 (L), Val 353 (L) | Ala 483 (L), Phe 383 (L), Lys 371 (L), Val 356 (L), Leu 348 (L) | Thr 386 (H), Leu 376 (L), Ile 251 (L), Val 259 (L), Ala 257 (L) | Leu 511 (L), Ala 521 (L), Met 458 (L), Lys 409 (H, L), Ala 407 (L), Val 394 (L), Phe 391 (L) |
| DihydroxyRoy (7) | Met 470 (L), Val 420 (H), Met 417 (L), Lys 368 (L), Leu 345 (H, L) | Phe 353 (L), Leu 348 (L), Val 356 (L), Lys 371 (L), Met 420 (R, L), Leu 394 (L), Phe 485 (L), Ala 483 (L) | Asp 373, Val 259, Lys 274, Ala 272, Ile 323 | Leu 511 (L), Ala 521 (L), Phe 523 (L), Leu 432 (L), Met 458 (L), Lys 409 (L), Val 394 (L), Leu 386 (L), Phe 391 (R) |
| PKCα | PKC βI | PKCι | PKCθ |
|---|---|---|---|
| Met 470 | Met 473 | Leu 376 | Leu 511 |
| Ala 480 | Ala 483 | Thr 386 | Ala 521 |
| Thr 401 | Thr 404 | Val 307 | Thr 442 |
| Met 417 | Met 420 | Ile 323 | Met 458 |
| Lys 368 | Lys 371 | Lys 274 | Lys 409 |
| Val 353 | Val 356 | Val 259 | Val 394 |
| Leu 345 | Leu 348 | Ile 251 | Leu 386 |
| Compound | Total Solvent Accessible Area (Å2) | Solved Exposed Area in Docked Pose (Å2) | Exposed Surface Ratio % | logP |
|---|---|---|---|---|
| DeRoy (1) | 268.47 | 142.36 | 53.02 | 4.53 |
| Roy (2) | 318.47 | 209.29 | 65.71 | 2.65 |
| ParvD (3) | 171.75 | 355.37 | 48.12 | 5.64 |
| RoyBz (4) | 469.79 | 260.01 | 55.35 | 7.88 |
| RoyBzCl (5) | 504.92 | 323.30 | 64.03 | 8.8 |
| RoyPr2 (6) | 403.77 | 207.97 | 51.50 | 4.87 |
| DihidroxyRoy (7) | 285.37 | 165.58 | 58.02 | 2.52 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isca, V.M.S.; Sencanski, M.; Filipovic, N.; Dos Santos, D.J.V.A.; Čipak Gašparović, A.; Saraíva, L.; Afonso, C.A.M.; Rijo, P.; García-Sosa, A.T. Activity to Breast Cancer Cell Lines of Different Malignancy and Predicted Interaction with Protein Kinase C Isoforms of Royleanones. Int. J. Mol. Sci. 2020, 21, 3671. https://doi.org/10.3390/ijms21103671
Isca VMS, Sencanski M, Filipovic N, Dos Santos DJVA, Čipak Gašparović A, Saraíva L, Afonso CAM, Rijo P, García-Sosa AT. Activity to Breast Cancer Cell Lines of Different Malignancy and Predicted Interaction with Protein Kinase C Isoforms of Royleanones. International Journal of Molecular Sciences. 2020; 21(10):3671. https://doi.org/10.3390/ijms21103671
Chicago/Turabian StyleIsca, Vera M. S., Milan Sencanski, Nenad Filipovic, Daniel J. V. A. Dos Santos, Ana Čipak Gašparović, Lucília Saraíva, Carlos A. M. Afonso, Patrícia Rijo, and Alfonso T. García-Sosa. 2020. "Activity to Breast Cancer Cell Lines of Different Malignancy and Predicted Interaction with Protein Kinase C Isoforms of Royleanones" International Journal of Molecular Sciences 21, no. 10: 3671. https://doi.org/10.3390/ijms21103671
APA StyleIsca, V. M. S., Sencanski, M., Filipovic, N., Dos Santos, D. J. V. A., Čipak Gašparović, A., Saraíva, L., Afonso, C. A. M., Rijo, P., & García-Sosa, A. T. (2020). Activity to Breast Cancer Cell Lines of Different Malignancy and Predicted Interaction with Protein Kinase C Isoforms of Royleanones. International Journal of Molecular Sciences, 21(10), 3671. https://doi.org/10.3390/ijms21103671