Denture Base Composites: Effect of Surface Modified Nano- and Micro-Particulates on Mechanical Properties of Polymethyl Methacrylate



Abstract
:1. Introduction
2. Aim
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Stage I—Polymerisation of MMA
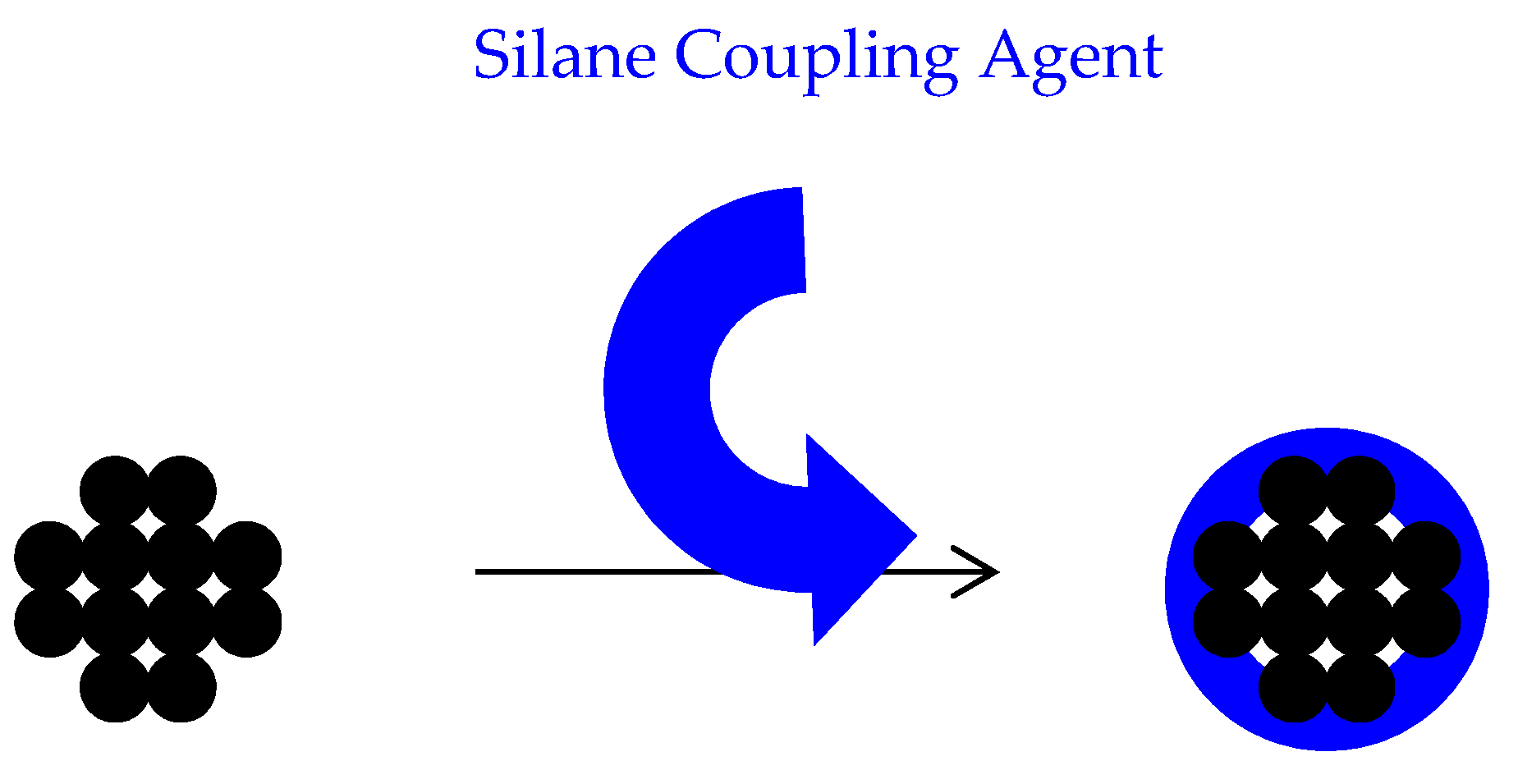
3.2.2. Stage II—Surface Modification of Particles
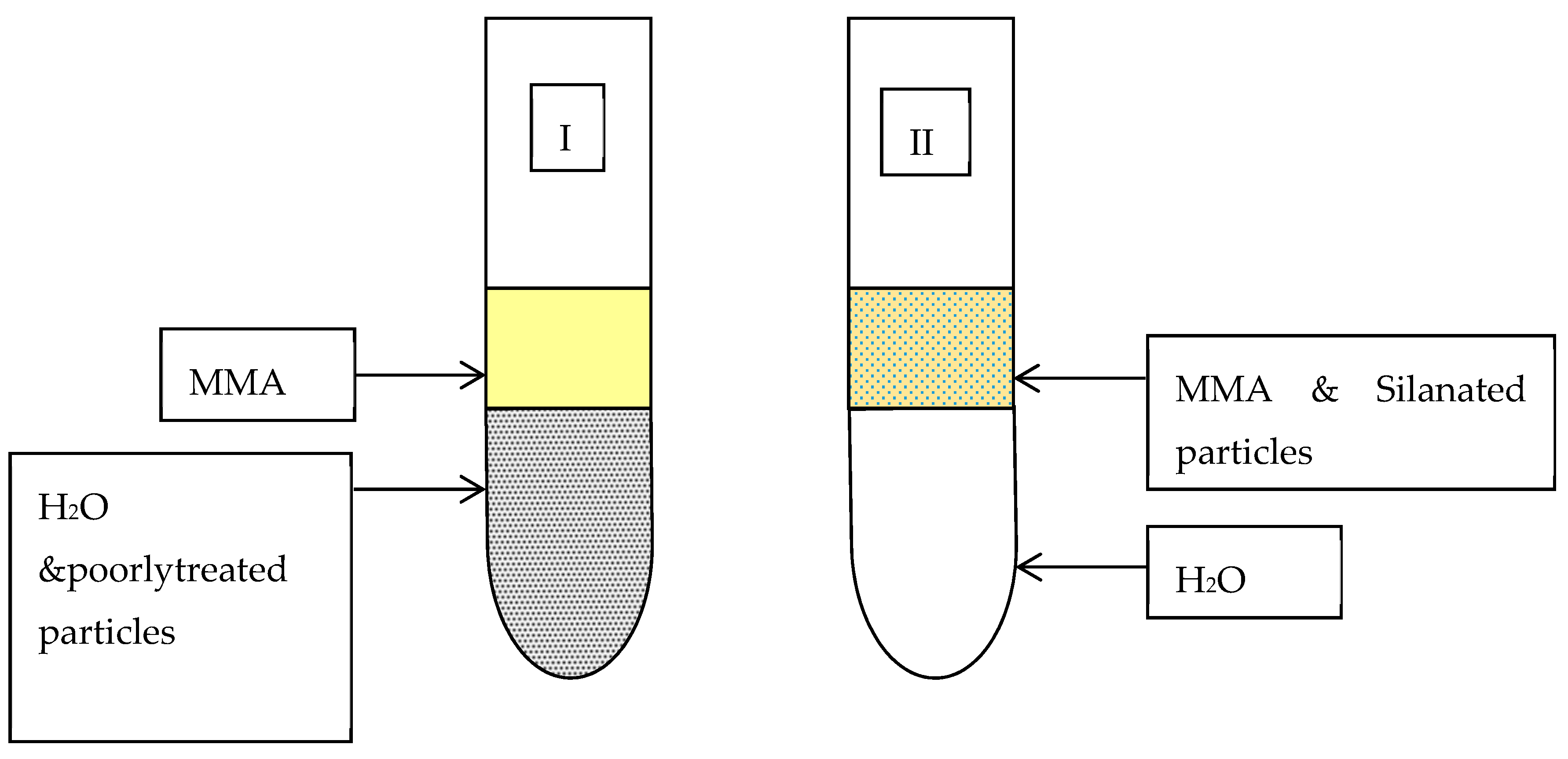
3.2.3. Stage III—Dispersion of Filler Particles within the Resin
3.3. Control Materials
3.4. Testing Method
ƒ (α/w) = 3 (α/w) 0.5 [ 1.99 − (α/w) (1 − α/w) (2.15 − 3.93 α/w + 2.7 α2/w2)] ÷ 2 (1 + 2 α/w) (1 − α/w)1.5
3.5. Statistical Analysis
4. Results
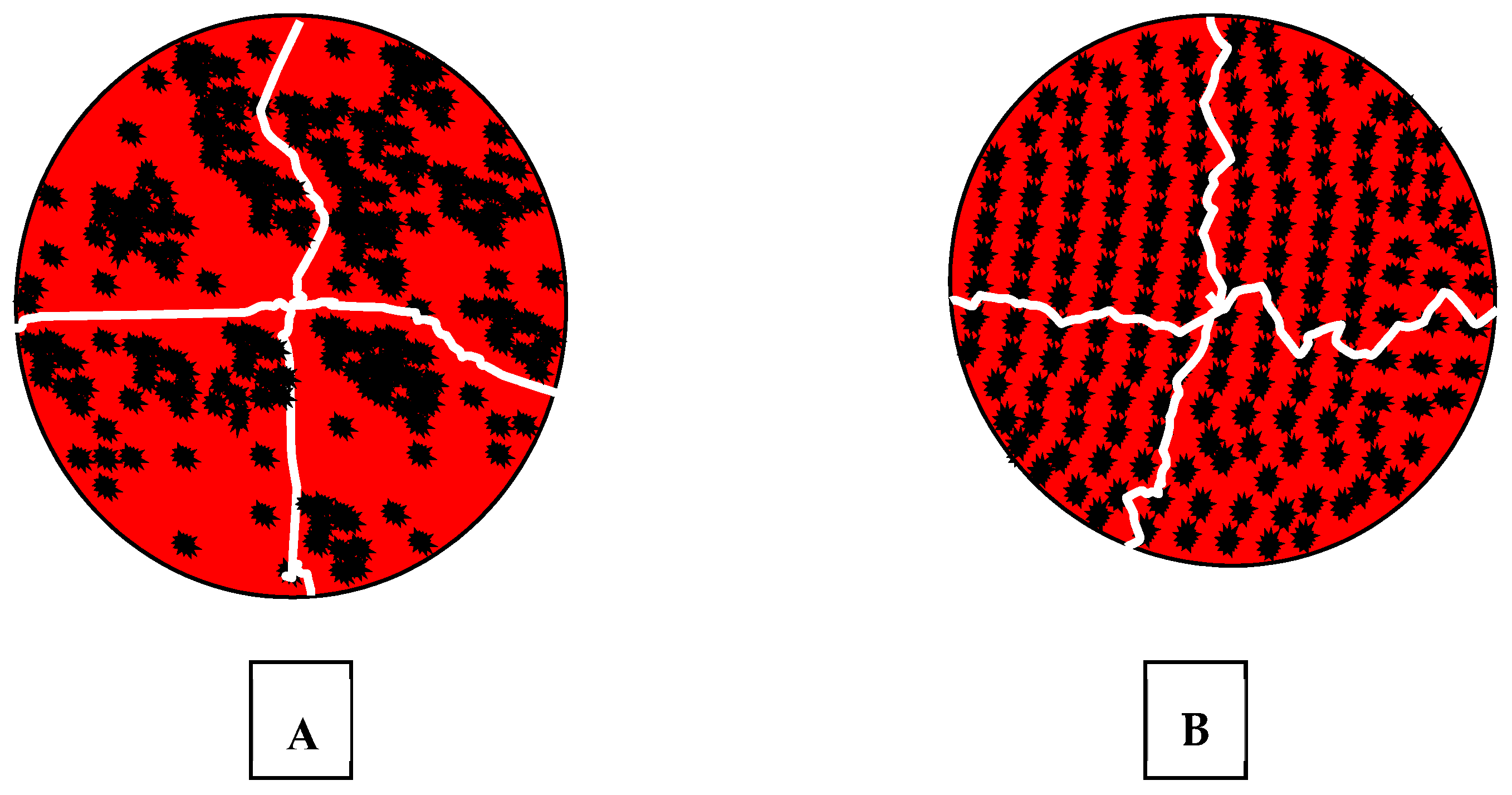
4.1. Surface Modification of Particles
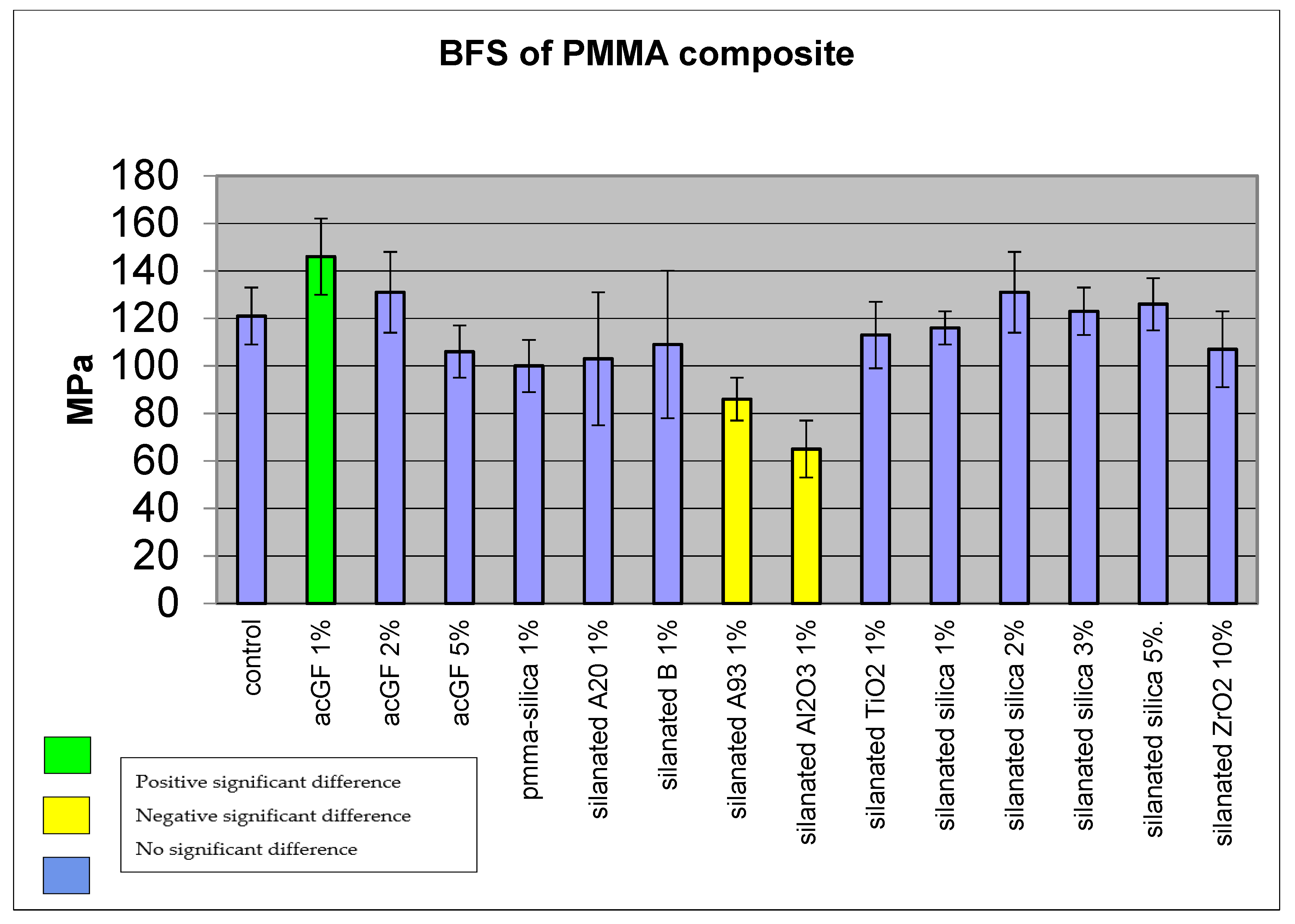
4.2. Biaxial Flexural Strength of Adapted Resins
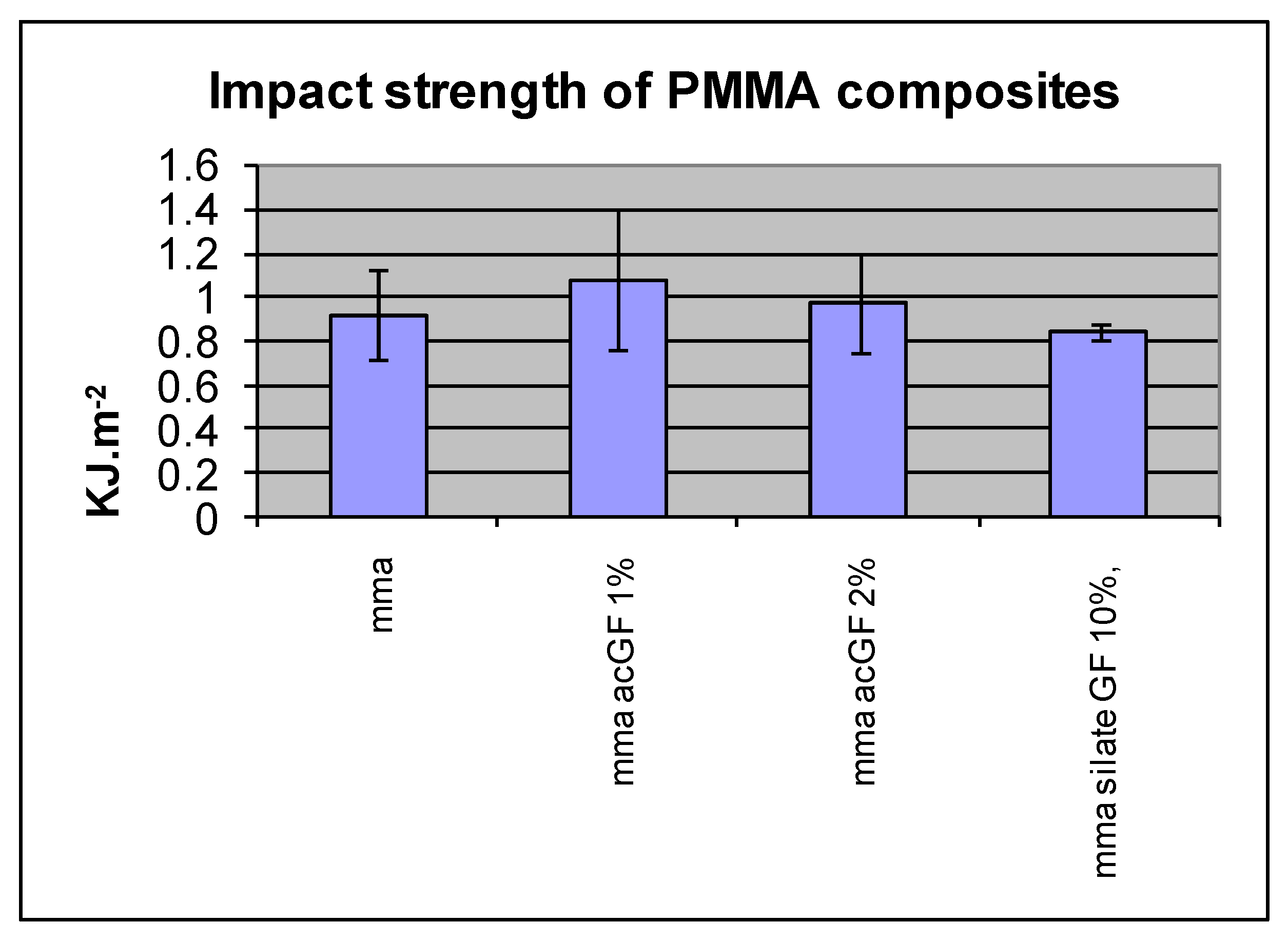
4.3. Impact Strength of Resins
4.4. Fracture Toughness of the Adapted Resins
4.5. High-Impact Resin Results
4.6. Impact Strength Results
4.7. Fracture Toughness Results
5. Discussion
5.1. Surface Treatment
5.2. Mechanical Properties of Filled Conventional Acrylic Resin
5.3. Mechanical Properties of High-Impact Resin
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Alla, R.; Raghavendra, K.N.; Vyas, R.; Konakanchi, A. Conventional and contemporary polymers for the fabrication of denture prosthesis: Part I—Overview, composition and properties. Int. J. Appl. Dent. Sci. 2015, 1, 82–89. [Google Scholar]
- Jagger, D.; Harrison, A.; Jandt, K. The reinforcement of dentures. J. Oral Rehabil. 1999, 26, 185–194. [Google Scholar] [CrossRef] [PubMed]
- John, J.; Gangadhar, S.; Shah, I. Flexural strength of heat-polymerized polymethyl methacrylate denture resin reinforced with glass, aramid, or nylon fibers. J. Prosthet. Dent. 2001, 86, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Darbar, U.; Huggett, R.; Harrison, A. Denture fracture—A survey. Br. Dent. J. 1994, 176, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Vallittu, P.; Alakuiala, P.; Lassila, V.; Lappalainen, R. In vitro fatigue fracture of an acrylic resin-based partial denture: An exploratory study. J. Prosthet. Dent. 1994, 72, 289–295. [Google Scholar] [CrossRef]
- Vallittu, P. Comparison of the in vitro fatigue resistance of an acrylic resin removable partial denture reinforced with continuous glass fibers or metal wires. J. Prosthodont. 1996, 5, 115–121. [Google Scholar] [CrossRef] [PubMed]
- El-Sheikh, A.M.; Al-Zahrani, S.B. Causes of Denture Fracture: A Survey. Saudi Dent. J. 2015, 18, 149–154. [Google Scholar]
- Härle, F. Follow-up investigation of surgical correction of the atrophic alveolar ridge by visor-osteotomy. J. Maxillofac. Surg. 1979, 7, 283–293. [Google Scholar]
- Ganzarolli, S.; De mello, J.; Shinkai, R.; Del Bel Cury, A. Internal adaptation and some physical properties of methacrylate-based denture base resins polymerized by different techniques. J. Biomed. Mater. Res. Part B Appl. Biomater. 2007, 82, 169–173. [Google Scholar] [CrossRef]
- Nejatian, T.; Sefat, F.; Johnson, T. Impact of Packing and Processing Technique on Mechanical Properties of Acrylic Denture Base Materials. Materials 2015, 8, 2093–2109. [Google Scholar] [CrossRef] [Green Version]
- Lamb, D.; Ellis, B.; Van Noort, R. The fracture topography of acrylic dentures broken in service. Biomaterials 1985, 6, 110–112. [Google Scholar] [CrossRef]
- Kim, S.; Watts, D. The effect of reinforcement with woven E-glass fibers on the impact strength of complete dentures fabricated with high-impact acrylic resin. J. Prosthet. Dent. 2004, 91, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Nejatian, T.; Johnson, A.; Van Noort, R. Reinforcement of denture base resin. Adv. Sci. Technol. 2006, 49, 124–129. [Google Scholar] [CrossRef]
- Stafford, G.; Huggett, R.; MacGregor, A.; Graham, J. The use of nylon as a denture-base material. J. Dent. 1986, 14, 18–22. [Google Scholar] [CrossRef]
- Yunus, N.; Rashid, A.; Azmi, L.; Abu-Hassan, M. Some flexural properties of a nylon denture base polymer. J. Oral Rehabil. 2005, 32, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Hedzelek, W.; Gajdus, P. Comparison of mechanical strength of palatal denture bases made from various plastic materials. Int. J. Prosthodont. 2006, 19, 193–194. [Google Scholar]
- Acquaviva, P.A.; Cerutti, F.; Adami, G.; Gagliani, M.; Ferrari, M.; Gherlone, E.; Cerutti, A. Degree of conversion of three composite materials employed in the adhesive cementation of indirect restorations: A micro-Raman analysis. J. Dent. 2009, 37, 610–615. [Google Scholar] [CrossRef]
- Tan, F.; Qiao, X.; Chen, J.; Wang, H. Effects of coupling agents on the properties of epoxy-based electrically conductive adhesives. Int. J. Adhes. Adhes. 2006, 26, 406–413. [Google Scholar] [CrossRef]
- Chmielewska, B.; Czarnecki, L.; Sustersic, J.; Zajc, A. The influence of silane coupling agents on the polymer mortar. Cement Concr. Compos. 2006, 28, 803–810. [Google Scholar] [CrossRef]
- Antonucci, J.; Dickens, S.; Fowler, B.; Xu, H.; Mcdonough, W. Chemistry of silanes: Interfaces in dental polymers and composites. J. Res. Natl. Inst. Stand. Technol. 2005, 110, 541–558. [Google Scholar] [CrossRef]
- Liu, Q.; Ding, J.; Chambers, D.; Debnath, S.; Wunder, S.; Bran, G. Filler-coupling agent-matrix interactions in silica/polymethylmethacrylate composites. J. Biomed. Mater. Res. 2001, 57, 384–393. [Google Scholar] [CrossRef]
- Praveen, B.; Babaji, H.V.; Prasanna, B.G.; Rajalbandi, S.K.; Shreeharsha, T.V.; Prashant, G.M. Comparison of Impact Strength and Fracture Morphology of Different Heat Cure Denture Acrylic Resins: An In vitro Study. JIOH 2014, 6, 12–16. [Google Scholar] [PubMed]
- Zidan, S.; Silikas, N.; Alhotan, A.; Haider, J.; Yates, J. Investigating the Mechanical Properties of ZrO2-Impregnated PMMA Nanocomposite for Denture-Based Applications. Materials 2019, 12, 1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albaladejo, A.; Osorio, R.; Papacchini, F.; Goracci, C.; Toledano, M.; Ferrari, M. Post silanization improves bond strength of translucent posts to flowable composite resins. J. Biomed. Mater. Res. Part B Appl. Biomater. 2007, 82, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Brentel, A.; Özcar, M.; Valandro, L.; Alarca, R.; Bottino, M. Microtensile bond strength of a resin cement to feldpathic ceramic after different etching and silanization regimens in dry and aged conditions. Dent. Mater. 2007, 20, 677–686. [Google Scholar] [CrossRef]
- Abboud, M.; Casaubieilh, L.; Morvan, F.; Fontanille, M.; Duguet, E. PMMA-based composite materials with reactive ceramic fillers: IV. Radiopacifying particles embedded in PMMA beads for acrylic bone cement. J. Biomed. Mater. Res. 2000, 53, 728–736. [Google Scholar] [CrossRef]
- British Standards Institution. BS7448-1. Fracture Mechanics Toughness Tests. Method for Determination of KIc, Critical CTOD and Critical J Values of Metallic Materials. 1991. Available online: https://shop.bsigroup.com (accessed on 8 January 2020).
- Piddock, V.; Marquis, P.M.; Wilson, H.J. Comparison of the strengths of aluminous porcelain fired on to platinum and palladium foils. J. Oral Rehabil. 1986, 13, 31–37. [Google Scholar] [CrossRef]
- Adabo, G.; Dos Santos Cruz, C.; Fonseca, R.; Vaz, L. The volumetric fraction of inorganic particles and the flexural strength of composites for posterior teeth. J. Dent. 2003, 31, 353–359. [Google Scholar] [CrossRef]
- Mortazavi, V.; Atail, M.; Fathi, M.; Keshavarzi, S.; Khalighinejad, N.; Badrian, H. The effect of nanoclay filler loading on the flexural strength of fiber-reinforced composites. Dent. Res. J. 2012, 9, 273–280. [Google Scholar]
- Halvorson, R.; Erickson, R.; Davidson, C. The effect of filler and silane content on conversion of resin-based composite. Dent. Mater. 2003, 19, 327–333. [Google Scholar] [CrossRef]
- Ihab, N.; Hassanen, K.; Ali, N. Assessment of zirconium oxide nano-fillers incorporation and silanation on impact, tensile strength and color alteration of heat polymerized acrylic resin. J. Baghdad Coll. Dent. 2012, 24, 36–42. [Google Scholar]
- Fu, J.; Naguib, H.E. Effect of Nanoclay on the Mechanical Properties of PMMA/Clay Nanocomposite Foams. J. Cell. Plast. 2006, 42, 325–342. [Google Scholar] [CrossRef]
- Shakeri, F.; Nodehi, A.; Atai, M. PMMA/double-modified organoclay nanocomposites as fillers for denture base materials with improved mechanical properties. J. Mech. Behav. Biomed. Mater. 2019, 90, 11–19. [Google Scholar] [CrossRef] [PubMed]
- MC Nally, L.; O’Sullivan, D.J.; Jagger, D.C. An in vitro investigation of the effect of the addition of untreated and surface treated silica on the transverse and impact strength of poly(methyl methacrylate) acrylic resin. Bio-Med. Mater. Eng. 2006, 16, 93–100. [Google Scholar]
- Anderson, T. Fracture Mechanics: Fundamentals and Applications; CRC Press: London, UK, 1995. [Google Scholar]
- Engineered Materials Handbook; ASM International: Cleveland, OH, USA, 1988.
- Cazzaniga, G.; Ottobelli, M.; Ionescu, A.C.; Paolone, G.; Gherlone, E.; Ferracane, J.L.; Brambilla, E. In vitro biofilm formation on resin-based composites after different finishing and polishing procedures. J. Dent. 2017, 67, 43–52. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PMMA Composite | BFS (MPa) | SD |
|---|---|---|
| Control (cured MMA) | 121 | 12 |
| ac GF, 1% | 146 * | 16 |
| ac GF, 2% | 131 | 17 |
| ac GF, 3% | 106 | 11 |
| PMMA-Silica, 1% | 100 | 11 |
| Closite A20, 1% | 103 | 28 |
| Closite B, 1% | 109 | 31 |
| A93, 1% | 86 * | 9 |
| Al2O3, 1% | 65 * | 12 |
| TiO2, 1% | 113 | 14 |
| Silica, 1% | 116 | 7 |
| Silica, 2% | 131 | 17 |
| Silica, 3% | 123 | 10 |
| Silica, 5% | 126 | 11 |
| ZrO2, 10% | 107 | 16 |
| PMMA Composite | IS KJ·m−2 | SD |
|---|---|---|
| Control (cured MMA) | 0.92 | 0.2 |
| ac GF, 1% | 1.08 | 0.32 |
| ac GF, 2% | 0.97 | 0.23 |
| ac GF, 3% | 0.84 | 0.03 |
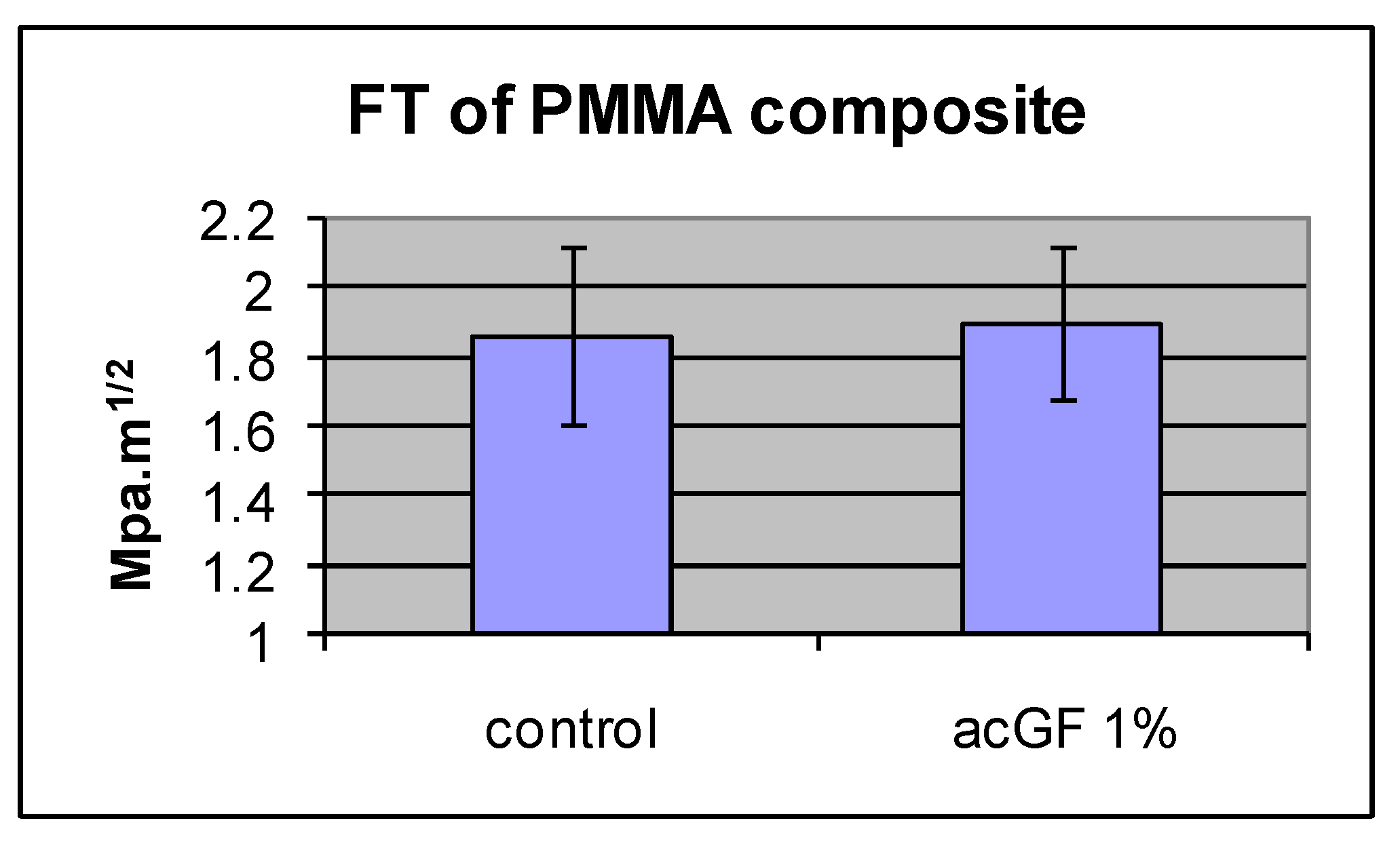
| PMMA Composite | FT MPa·m1/2 | SD |
|---|---|---|
| Control (cured MMA) | 1.86 | 0.25 |
| ac GF, 1% | 1.9 | 0.22 |
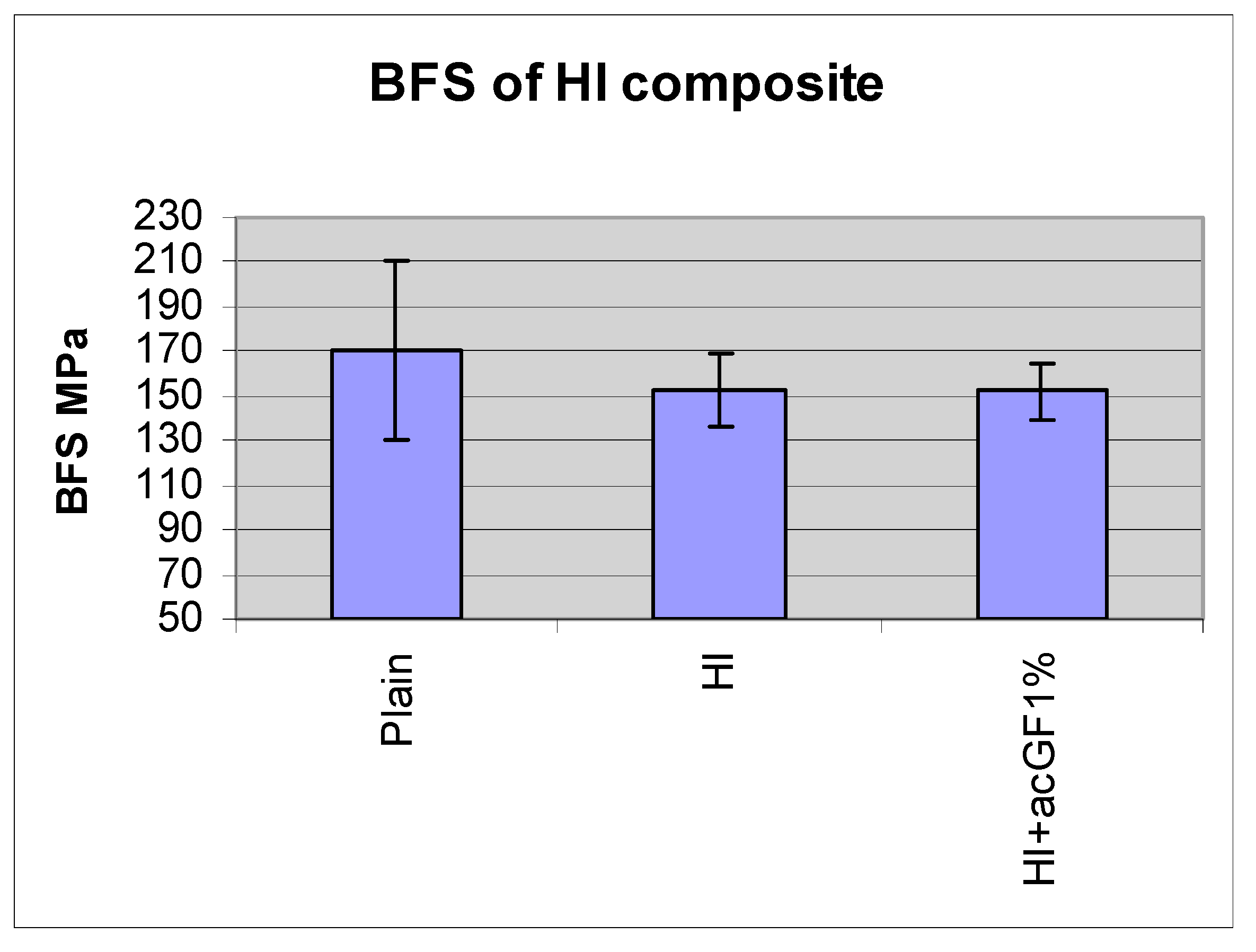
| Plain | HI | HI+acGF1% | |
|---|---|---|---|
| BFS MPa | 170 | 153 | 152 |
| SD | 40 | 16 | 13 |
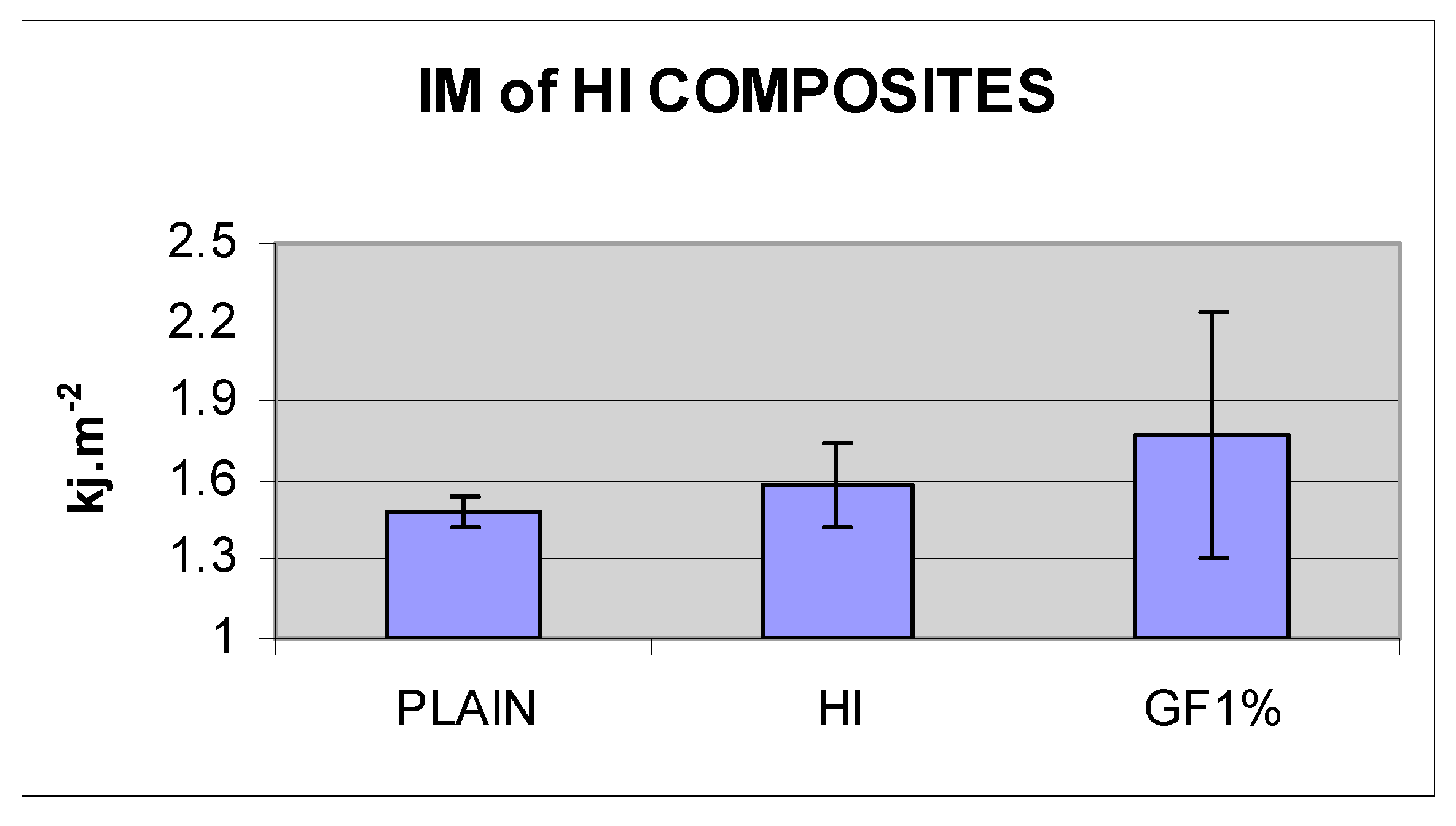
| PLAIN | HI | HI+GF1% | |
|---|---|---|---|
| IM KJ·m−2 | 1.48 | 1.58 | 1.77 |
| SD | 0.06 | 0.16 | 0.47 |
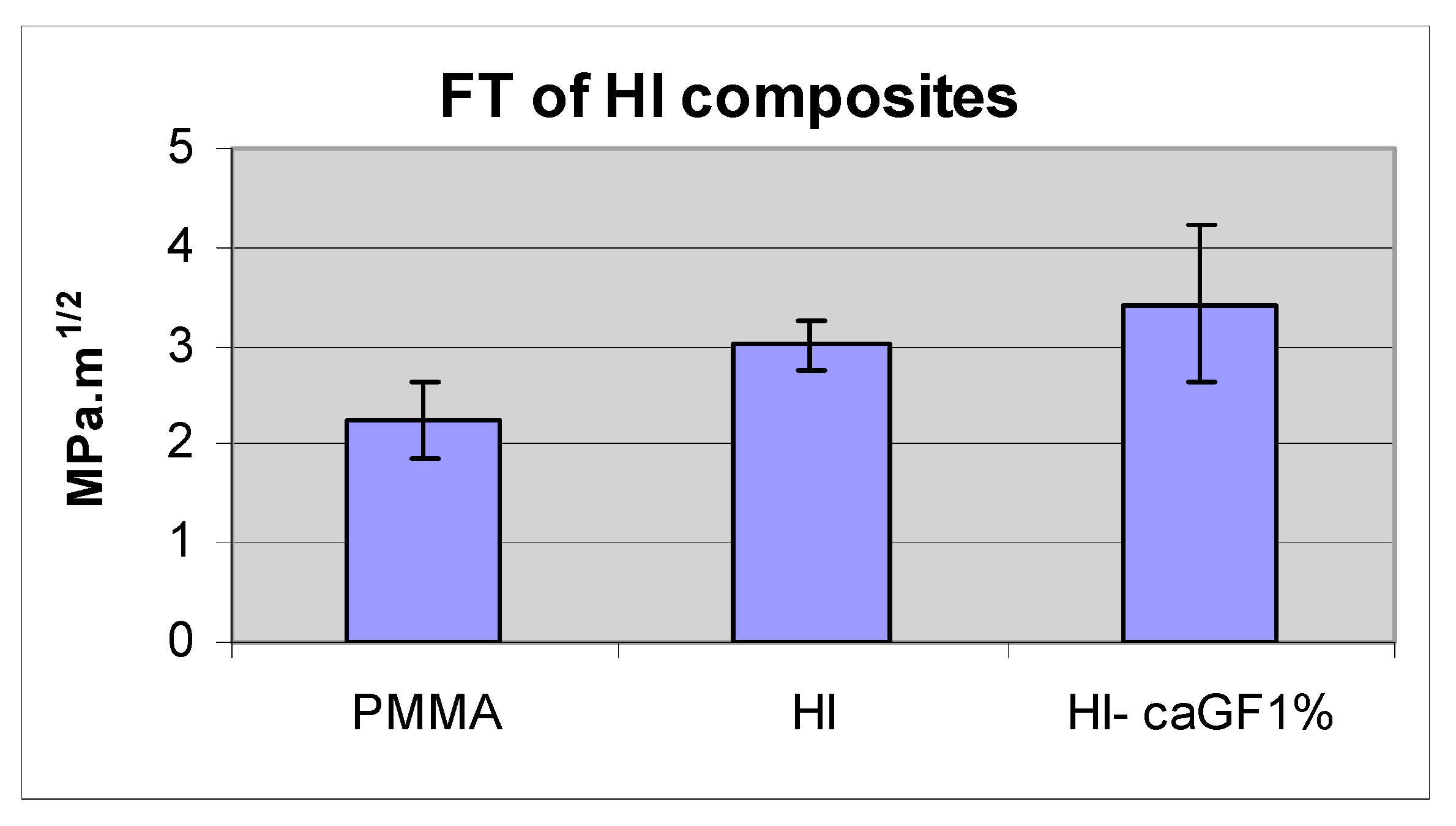
| PMMA | HI | HI+GF1% | |
|---|---|---|---|
| FT MPa·m1/2 | 2.24 | 3.02 * | 3.43 |
| SD | 0.38 | 0.25 | 0.78 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nejatian, T.; Nathwani, N.; Taylor, L.; Sefat, F. Denture Base Composites: Effect of Surface Modified Nano- and Micro-Particulates on Mechanical Properties of Polymethyl Methacrylate. Materials 2020, 13, 307. https://doi.org/10.3390/ma13020307
Nejatian T, Nathwani N, Taylor L, Sefat F. Denture Base Composites: Effect of Surface Modified Nano- and Micro-Particulates on Mechanical Properties of Polymethyl Methacrylate. Materials. 2020; 13(2):307. https://doi.org/10.3390/ma13020307
Chicago/Turabian StyleNejatian, Touraj, Neil Nathwani, Louise Taylor, and Farshid Sefat. 2020. "Denture Base Composites: Effect of Surface Modified Nano- and Micro-Particulates on Mechanical Properties of Polymethyl Methacrylate" Materials 13, no. 2: 307. https://doi.org/10.3390/ma13020307
APA StyleNejatian, T., Nathwani, N., Taylor, L., & Sefat, F. (2020). Denture Base Composites: Effect of Surface Modified Nano- and Micro-Particulates on Mechanical Properties of Polymethyl Methacrylate. Materials, 13(2), 307. https://doi.org/10.3390/ma13020307