1. Introduction
Dearomatization has become an important subject in environmental catalysis worldwide to obtain clean fuels. The presence of aromatics in fuel reduces the cetane number in diesel and enhances the emission of particulate matter which is known to be responsible for various health problems when inhaled. Therefore, environmental regulations are directed to decrease these emissions from vehicle exhausts. Moreover, the aromatic reduction of 10 wt.% results in cetane number increase by ~3 allowing better engine operation [
1]. The removal of aromatic compounds from gasoline or diesel is realized by catalytic hydrogenation [
2].
Noble metal catalysts, which work at relatively low temperatures, are important for clean fuel production by aromatic saturation. However, these catalysts are very sensitive to the sulfur- and nitrogen-containing compounds presented in the feedstock [
3]. The hydrodearomatization activity and sulfur-tolerance of noble-metals catalysts may be enhanced by deposition of noble metal (Pt, Pd, etc.) on acidic supports such as zeolite H-ZSM-5, ultrastable zeolite Y (USY), H-beta, mordenite, or alumina [
4,
5,
6,
7]. These effects are caused by the synergistic effects between noble metal particles and acid sites of support. The electron transfers partially from the metal particles to the acid sites that lead to the formation of electron-deficient metal (Me
δ+), which decreases the strength of the sulfur-metal bond preventing the formation of metal sulfides and the noble metal particles (active sites) become more stable [
8]. Moreover, the acid sites of the support provide additional active sites on which aromatic and sulfur compounds are hydrogenated by the spillover hydrogen from the metal surface [
9]. It was shown that noble metals supported on zeolites are more active than supported on alumina. On the other hand, a high acidity of zeolites also favors excessive cracking and coke formation causing quick deactivation of the catalysts [
10,
11]. No deactivation was observed when the metals were supported on alumina or SiO
2–TiO
2, the supports with weak and intermediate acid sites [
12,
13].
It was shown that treatment of siliceous zeolites with a MFI structure as well as amorphous silica with an aqueous solution of ammonium salts and at least one basic compound generates their additional acidity [
14,
15,
16,
17]. The Fourier-transform infrared spectroscopy (FT-IR) indicated the creation of H-bonded silanol defect groups of different kinds with some acidity in the samples modified in this way. It caused also the generation of mesopores by partial leaching of silicon from the framework during the modification procedure [
16]. It allows decreasing the diffusion limitation if they are used as catalysts or support of catalysts.
Within the above background, the purpose of this work was to use the silicalite materials with MFI structure (silicalite-1) with different acidity as supports of iridium catalysts. The acidic sites in pristine silicalite-1 were generated employing post-synthesis treatment with solutions of various ammonium agents (NH
4F, NH
4OH) at elevated temperature [
16]. The influence of support acidity on the efficiency of the resulting iridium catalysts for toluene hydrogenation was examined. Toluene was used as a model compound to simulate the aromatics in diesel fuels because the hydrogenation of toluene is known to be more difficult than that of benzene and naphthalene [
18]. Taking into account the influence of texture and the acid function of supports on the activity of such catalysts, the characterization of the supports and catalysts was performed using Brunauer–Emmet–Teller (BET) method, temperature-programmed reduction with hydrogen (TPR-H
2), H
2 chemisorption, transmission electron microscopy (TEM), temperature-programmed desorption of ammonia (TPD-NH
3), and X-ray photoelectron spectroscopy (XPS). The catalytic activity of iridium catalysts supported on unmodified and modified silicalite-1 was compared. The impact of texture and acidity of catalysts on the activity of iridium catalysts was assessed in the reaction of toluene hydrogenation. To the best of our knowledge, no data are available on such modifications of silicalite-1 followed by the application of resulting material as iridium catalyst support for hydrogenation processes.
2. Materials and Methods
Silicalite-1 (denoted as Sil) was prepared according to our earlier procedure [
16]. The calcined silicalite-1 was modified using 1 M solutions of various ammonium compounds (NH
4OH or NH
4F, Aldrich, Saint Louis, MO, USA). The sample (1 g) of silicalite-1 was mixed with the respective NH
4+ source solution (100 cm
3). The mixture (the silicalite-1 and aqueous solution of ammonium compounds) was stirred under reflux at 60 °C for 1 h. After the treatment, the samples were filtered, washed with deionized water, dried at 105 °C for 24 h, and then calcined in air (Linde, Pullach im Isartal, Germany) at 550 °C for 3 h with a temperature ramp of 5 °C⋅min
−1. The resulting samples were labelled as Sil-X, where X stands for the anion (OH
− or F
−) of applied ammonium compounds (e.g., SilOH was prepared with 1 M NH
4OH solution). Removal of fluorine and decomposition of ammonium cations in modified silicalite-1 was confirmed by means of energy dispersive X-ray spectroscopy (EDS analysis) (Thermo Fisher Scientific, Waltham, MA, USA, with an Ultra Dry Silicon Drift X-ray Detector), X-ray photoelectron spectra (XPS, Ultra High Vacuum (UHV) System (Specs, Berlin, Germany)), and X-ray fluorescence (XRF, Mini-Pal 2 spectrometer (Panalytical, Malvern, Worcestershire, UK)).
The Ir/support (support = Sil, Sil-OH or Sil-F; content of Ir was 1 wt.%) catalysts were prepared by the conventional impregnation method using hexachloroiridium (IV) acid hydrate (H2IrCl6⋅× H2O, 99.995%, Aldrich, Saint Louis, MO, USA) as a metal precursor. An appropriate amount of support was placed in an aqueous solution of H2IrCl6⋅× H2O followed by evaporation on a rotary evaporator (Heidolph, Schwabach/Nuremberg, Germany). Then the supports loaded with precursors were dried at 105 °C for 24 h and labelled as Ir/Sil-D, Ir/Sil-OH-D, and Ir/Sil-F-D.
Before measurements of hydrogen chemisorption, low-temperature nitrogen adsorption-desorption (BET surface area and porosity), determination of metal content (inductively coupled plasma optical emission spectrometry, ICP-OES), temperature-programmed desorption of ammonia (TPD-NH3), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), and catalytic activity, precursor-impregnated supports (Ir/Sil-D, Ir/Sil-OH-D, and Ir/Sil-F-D) were reduced in hydrogen flow (99.99%, Linde, Pullach im Isartal, Germany, 50 cm3⋅min−1) with a temperature ramp of 10 °C⋅min−1. After reaching the setpoint (400 °C), the catalysts reduction was continued for 2 h. The reduced catalysts were labelled as Ir/Sil, Ir/Sil-OH, and Ir/Sil-F.
The Brunauer–Emmet–Teller (BET) surface areas were determined by N2 adsorption-desorption at −196 °C using a Micromeritics ASAP 2010 sorptometer (Micromeritics, Norcross, GA, USA). Total pore volume and average pore diameter were determined by BET method and by applying the Barrett–Joyner–Halenda (BJH) method to the desorption branch of the isotherm. The microporous and mesoporous surface area were determined from t-plot method. Prior to the measurements, the samples were outgassed at 225 °C.
Powder X-ray diffraction patterns of supports and catalysts were recorded on the Philips Bruker D8 Advance diffractometer (Billerica, MA, USA) using Cu Kα radiation (λ = 1.54056 Å).
The metal loading in the catalysts after reduction (H2; 400 °C; 2 h) was determined by ICP-OES method (Inductively Coupled Plasma Optical Emission Spectroscopy) on a Varian Vista-MPX spectrometer (Palo Alto, CA, USA).
Measurements of temperature-programmed reduction with hydrogen (TPR-H
2) were carried out on Pulse ChemiSorb 2705 (Micromeritics, Norcross, GA, USA) instrument. The hydrogen consumption was monitored with a thermal conductivity detector (TCD) and the signal was normalized to the same sample weight—100 mg. Detailed experimental procedure of TPR-H
2 analysis is presented in the
Supplementary Materials.
The temperature-programmed desorption of ammonia (TPD-NH
3) measurements were carried out on Pulse ChemiSorb 2705 instrument (Micromeritics, Norcross, GA, USA) for the catalysts reduced at 400 °C for 2 h. The TPD-NH
3 measurements of acidity were performed in a flow reactor. Detailed experimental procedure of TPD-NH
3 analysis is presented in the
Supplementary Materials.
Transmission electron microscope (TEM) images were recorded on a JEOL 2000 microscope (Akishima, Tokyo, Japan) operating at accelerating voltage of 80 kV. The average particle was calculated from 100 particles using ImageJ program (v.1.53e, 2020, National Institutes of Health and the Laboratory for Optical and Computational Instrumentation (LOCI, University of Wisconsin, Madison, WI, USA)).
The hydrogen chemisorption measurements were conducted by the static method at 35 °C on an ASAP 2010C sorptometer (Micromeritics, Norcross, GA, USA). Detailed experimental procedure of H
2 chemisorption analysis is presented in [
19] and in the
Supplementary Materials.
X-ray photoelectron spectroscopy (XPS) analysis of supports and catalysts were carried out with an ultra-high vacuum (UHV) system (Specs, Berlin, Germany). The examined materials were irradiated with a monochromatized aluminum X-ray source (Al Kα (1486.6 eV)). The charge referencing method used was the C (C, H) component of the C 1s peak of adventitious carbon fixed at 284.6 eV. Spectroscopic data were processed by the CasaXPS ver. 2.3.17PR1.1 software (2016, Casa Software Ltd., Teignmouth, UK), using a peak-fitting routine with Shirley background.
Toluene hydrogenation was performed at atmospheric pressure using a fixed-bed flow reactor and H
2 as carrier gas. Before the reaction was started, fresh catalyst (25 mg) was placed into the reactor and reduced in situ in a flow (100 cm
3⋅min
−1) of pure hydrogen (99.99%) at 400 °C for 2 h. The reaction mixture was prepared by passing hydrogen (50 cm
3⋅min
−1) through a saturator filled with toluene (Aldrich, Saint Louis, MO, USA) and the obtained gaseous mixture (toluene and hydrogen) was directed to the reactor. The concentration of toluene in the feed was stable and established to 0.75 μmol⋅cm
−3. The catalytic activities were measured at temperatures range of 75–225 °C. Details of the hydrogenation reaction were presented in [
17]. The scheme (
Figure S1) of the setup used for toluene hydrogenation and detailed experimental procedure are also presented in
Supplementary Materials. For selected samples, a stability test at 125 °C for 22 h was also performed. The products were analyzed every 10 min by a SRI gas chromatograph (SRI Instruments, Earl St. Torrance, CA, USA) equipped with a capillary column Restek MXT-1 and a TCD detector. The catalytic activity was expressed as an apparent rate calculated by the following equation [
19]:
(where
F—total flow rate of feed (cm
3⋅min
−1);
Y—fractional conversion;
C—concentration of toluene in the feed (mol
Tl⋅cm
−3); and
N—iridium content (mol
Ir) in the sample) or as turnover frequency TOF (min
−1) in moles of toluene reacted per surface Ir atoms (determined by hydrogen chemisorption).
3. Results
X-ray diffractograms of the supports and catalysts (
Figure S2) confirmed the correct MFI structure of the support [
16]. The crystallinity of the catalysts is comparable to the crystallinity of the supports. Characteristic peaks of metallic Ir species (2θ = 40.6° (111) and 47.3° (200)) were not observed due to the low iridium content (1 wt.%) as well as a high dispersion of iridium species on the supports [
20].
One of the important parameters characterizing micro- and mesoporous systems is their BET surface area and porosity. The textures of supports after calcination at 550 °C (
Figure 1A,B,
Table 1) and iridium catalysts after reduction at 400 °C (
Figure 1C,D,
Table 1) were characterized by the low-temperature nitrogen adsorption-desorption measurements.
The N
2 adsorption-desorption curves of unmodified Sil are typical for Silicalite-1 materials presented in the literature [
16,
21] and correspond to type Ib (according to the International Union of Pure and Applied Chemistry (IUPAC) classification) characteristic for materials having pore size distributions over a broader range including wider micropores and possibly narrow mesopores (<2.5 nm) (
Figure 1A,B,
Table 1) [
22]. The low-temperature N
2 sorption isotherms exhibit two hysteresis loops. The first one at a low partial pressure p/p
0 = 0.1–0.4 range is detected for highly silica and pure silica MFI materials and can be attributed to the defects in the structure [
23]. The increasing volume of this hysteresis loop for modified samples can be explained by the formation of the additional framework defects as a result of the modification. The second hysteresis loop at p/p
0 > 0.5 for the Sil is attributed to the inter-crystalline porosity. The modification of silicalite-1 with ammonium compounds solutions leads to an increase of the BET surface area for all supports regardless of the type of used ammonium agents. The increase of the total pore volume and average pore diameter was observed especially after the use of NH
4OH—
Table 1. This increase is due to the formation of an additional mesoporous structure. The pore volume of the Sil-OH sample is one and a half times higher in comparison to the pore volume of the unmodified sample. The growing second loop in isotherms of the modified samples can be explained by a generation of additional mesoporosity during the used procedure. As shown in
Figure 1B, some mesoporosity, resulted from the inter-crystalline voids, is seen in the pristine silicalite-1 (Sil), whereas the modified samples show the presence of additional, well-defined pores with larger diameters compared to those of the starting material. The average pore sizes calculated by the BJH method are larger for modified supports and are 8.4 and 5.7 nm for Sil-OH and Sil-F samples, respectively—
Figure 1. The low-temperature nitrogen adsorption-desorption isotherms were also used to calculate surface area, cumulative pore diameter, and cumulative pore volume of iridium catalysts—
Table 1. During the preparation of catalysts, by the impregnation method, the active phase covers the surface of the support. Due to the low amount of introduced active phase, the impregnation with iridium precursor does not change the texture of the supports—
Figure 1C,D. The BET surface area, pore volume, and average pore size are similar to these values obtained for the supports before impregnation (
Table 1). The nitrogen adsorption isotherms (
Figure 1C) and pore size distribution (
Figure 1D) of the catalysts are almost similar to those of the supports. Only the surface of the micropores and mesopores changed. A decrease is observed in the microporous surface area with simultaneous increase of the mesoporous surface area in comparison to their values for supports. It may be related to the blocking of the smallest channels by the crystallites of the introduced active phase as well as the formation of additional mesoporosity by treatment with acidic iridium source—
Figure 2. The decrease of the volume of low-partial pressure hysteresis loop for iridium catalysts can indicate that some parts of the defect sites participate in the deposition of the active phase on the surface of supports.
TPD-NH
3 analysis allows determining not only the acid center concentration (
Table 1) on the support or catalyst surface, but also estimating the distribution of their acid strength (
Table 2). The TPD-NH
3 profiles of the obtained catalysts and the results of their deconvolution were presented in
Figure 3 and
Table 2. The shape of the TPD-NH
3 profiles indicates the presence of acid centers of different strengths—weak, medium, and strong. The desorption peaks at 265–310 °C range indicate the presence of weak centers, the peak with the maximum at about 360 °C corresponds to medium-strength centers, whereas from strong centers ammonia desorbed over 360 °C [
24]. The maximum desorption temperature in our study was just 400 °C what was dictated by the temperature of the activation of iridium catalysts. Our previous studies (TPD-NH
3 studies of supports [
16]) showed a low acidity of the starting material. The TPD-NH
3 profiles of the modified samples indicated the formation of additional acid sites as a result of the modification procedure. The number and the strength of created acid sites depended on the type of ammonium agents used for modification—
Table 1.
The sample modified with NH
4F indicated the highest overall acidity and the higher contribution of strong acid centers in comparison to the other samples. The sample modified with NH
4OH shows lower total acidity in comparison to the acidity of Sil-F with a predominance of weak and moderate acid sites. The TPD-NH
3 profiles of iridium catalysts allowed to calculate the concentration of surface acid centers of different strength (
Table 2). The TPD-NH
3 analysis indicated that the introduction of iridium phase on the surface of unmodified and modified silicalite-1 leads to a slight increase of the total acidity. The increase of total acidity of the supported noble metal catalysts in comparison to the acidity of the supports has been previously presented in the literature in the case of using metal chlorides or chlorometallic acids as a metal precursor. It was shown that the introduction of chlorides produced new acid sites [
25].
The formation of defects playing the role of acid centers is also confirmed by the XPS measurements of the supports before and after the modification with ammonium compounds. In the Si 2p spectrum of the initial silicalite-1, only one signal was observed at binding energy BE = 103.55 eV. Modification with NH
4OH as well as NH
4F leads to an additional weak signal at a lower energy of ~101 eV. According to Alfonsetti et al. [
26], this signal corresponds to SiO
x, where x < 2. It indicates the partial removal of oxygen from the framework of silicalite-1 during modification and was confirmed by the calculated amount of oxygen and by the Si/O atomic ratio, which increases from the value of 0.66 for Sil by 0.67 for Sil-OH to 0.73 for Sil-F (
Table 3). The formation of SiO
x species (x < 2) is also confirmed by the presence of an additional O 1s signal at BE~530.7 eV in the spectra of modified silicalites. The elimination of oxygen from the silicalite-1 framework results in the generation of oxygen vacancies, i.e., coordinated unsaturated silicon atoms that can act as Lewis acid centers.
To obtain information on the reducibility of active phase precursors introduced into the supports studied, temperature-programmed reduction with hydrogen (TPR-H
2) was performed. TPR-H
2 measurements were carried out on fresh catalysts dried at 105 °C. No reduction signals were seen in profiles of the supports (data not shown), which indicates the irreducibility of the supports. TPR-H
2 profiles of catalysts are presented in
Figure 4.
For the fresh catalyst precursors, two regions of reduction were observed at 125–250 °C and 250–475 °C. Preliminary analysis of these profiles shows that maxima of reduction of active phase precursors deposited on inert silicalite-1 and modified silicalite-1 occurred at different temperatures and the peak intensities depended on the type of supports used. This is a result of differences in the strength of the interaction between precursors and the surface of the above supports that can influence the dispersion of precursor on the supports.
The TPR-H
2 profiles of Ir/Sil-D catalysts show the main peak in the 125–250 °C range. This peak can be attributed to the reduction of highly dispersed iridium species coming from the reduction of precursor to metallic iridium [
19,
27,
28]. The degree of reduction of the iridium precursor calculated on the basis of hydrogen consumption was 77.6% for the peak with a maximum at 173 °C (
Table 4). In the case of catalysts supported on modified silicalite-1 supports the main peak is also present in the 125–250 °C range, but the maximum of peaks is shifted to higher temperatures. For comparison, the TPR-H
2 studies of iridium precursor impregnated on quartz sand was done showing that such supported hydrogen hexachloroiridate is reduced at 190 °C—inset in
Figure 4. The absence of the reduction peaks above 245 °C in the TPR-H
2 profile of the precursor (H
2IrCl
6) indicates the differences in metal-support interactions in comparison to interaction for investigated catalysts as well as the absence of larger clusters of iridium supported on quartz sand (
Figure 4, inset). The only peak at 190 °C is observed in the reference sample, which indicates that the iridium precursor is fully reduced at this temperature [
29]. The signals in the 245–475 °C region present in the TPR-H
2 profiles of investigated catalysts are assigned to larger clusters of iridium or to iridium stronger bonded to the support surface [
19]. The reduction of the iridium phase supported on both modified silicalite-1 in this region takes place at a lower temperature in comparison to the temperature of reduction for Ir/Sil-D. The maxima of reduction are shifted to lower temperature in the following order Ir/Sil-F-D < Ir/Sil-OH-D < Ir/Sil-D and they are 343, 369, and 398 °C, respectively. It indicates that the temperature used to activate the catalysts (pure H
2, 400 °C, 2 h) is sufficient for the reduction of the active phase precursor to the metallic phase. The degree of reduction at the higher temperature range depends on the type of supports used and is the lowest for Ir/Sil-D (22.2%) and the highest for Ir/Sil-F-D sample (34.1%) with the highest contribution of medium and strong acid sites. The above-presented data indicates that the contribution of the high-temperature peak of reduction increases with the increasing acidity of the support used (
Table 1 and
Table 2 and
Figure 3). A similar observation was presented by D’Ippolito et al. [
25] for Ir/Al
2O
3-SiO
2 systems. The TPR-H
2 data are in good agreement with the N
2 adsorption/desorption results (decrease of the volume of low-partial pressure hysteresis loop) indicating the interaction of iridium precursor with the formed defects acting as acid sites.
Another important parameter characterizing metallic catalysts is the dispersion of the active phases. The amount of chemisorbed hydrogen permits the determination of the average size of Ir particles and also the number of iridium surface atoms that is required for the calculation of turnover frequency (TOF, min
−1) of toluene hydrogenation—
Table 5.
The results of hydrogen chemisorption measurements indicate that the textural and acidic properties of supports have an influence on the dispersion and particle size of the iridium active phase. The Ir/Sil-F and Ir/Sil-OH catalysts show much higher dispersion than catalyst supported on unmodified silicalite-1 (
Table 5). The highest dispersion was observed for Ir/Sil-OH catalyst (D~34%) with the highest surface area and pores size as well as with high acidity of medium strength. This catalyst exhibits slightly higher dispersion and lower particle size (3.3 nm) when compared to iridium catalyst supported on Sil-F (D~29%, Ir particle size = 3.8 nm). The last one has a higher contribution of stronger acid sites, in comparison to the Sil-OH support, that interacts stronger with iridium precursor causing the formation of bigger Ir particles. Using the modified silicalite-1 supports with mesoporous structure allows to attain higher dispersion and lower particle size compared to iridium catalysts supported on pristine silicalite-1 support, where dispersion was ~22% and Ir particle size ~5 nm. It indicates that the mesoporous structure and large surface area of the modified supports have a crucial effect on the metal dispersion. The acidity of the support has a less significant influence on dispersion and the size of iridium particles.
The particle size was also estimated by TEM analysis—
Figure 5. The TEM images and particle size distribution histograms show that the iridium crystallite size for Ir/Sil (
Figure 5A) is almost 50% larger than for the iridium particles supported on the modified silicalite-1 samples. The presented pictures also confirm the homogeneous dispersion of the nanoparticles on the surface of all supports.
The TPD-NH
3 studies for our supports (Sil-F and Sil-OH—
Table 1) indicated a higher number of stronger acid centers for Sil-F material. The lower acidity of the Sil-OH surface favors a slightly better dispersion of the iridium active phase. A similar observation was presented in the literature for modified amorphous silica [
17]. The fact that iridium species are weakly bonded to the unmodified support due to its low acidity must also be considered. The presented H
2 chemisorption results are in agreement with TPR-H
2 data showing the lowest reduction degree in the 245–475 °C range for Ir/Sil catalyst (
Table 5). As a consequence, iridium species show easy migration during the reduction step of the catalyst preparation by forming bigger iridium particles [
30].
The XPS analysis of the Ir 4f spectra of reduced iridium catalysts supported on modified silicalite-1 is shown in
Figure 6. The Ir 4f spectra can be fitted into one doublet. The peaks at ~61 and ~64 eV for Ir 4f
7/2 and Ir 4f
5/2 were assigned to iridium in the zero valent state [
31,
32]. There are no peaks at ~62 and ~65 eV attributed to the binding energies of the high valence state of iridium [
32,
33,
34]. It indicates that all iridium phase is reduced to the metallic form.
The catalytic activity of the iridium catalysts has been tested in the hydrogenation of toluene. As we mentioned before, the amount of chemisorbed hydrogen permits to determine the number of iridium surface atoms that is required for the calculation of turnover frequency (TOF, min
−1) of toluene hydrogenation.
Figure 7 shows the dependence of apparent rate (r
t) and TOF on iridium dispersion. Calculations of r
t and TOF have shown that the most active catalyst was Ir/Sil-OH, whose activity was almost 35% higher than that of Ir/Sil-F and was more than 2.5 times higher than the activity of Ir/Sil. The activity of the investigated catalysts, presented as r
t as well as TOF, increased with the dispersion of the active phase.
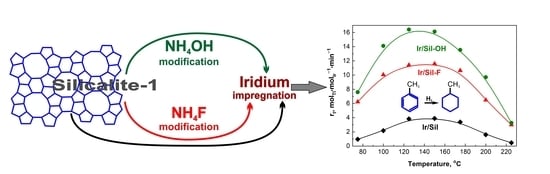
The results of the hydrogenation of toluene performed at different temperatures are presented in
Figure 8. The catalytic measurements carried out on the supports alone proved no activity in the hydrogenation of toluene.
The activity of the iridium catalyst supported on silicalite-1 modified with NH
4OH expressed as the apparent rate (r
t) of reaction (
Figure 8) was higher in comparison to the activity obtained over the catalyst supported on Sil-F and unmodified silicalite-1 in all investigated temperatures. The activity increased with the rise of the reaction temperature reaching a maximum at 125 °C for Ir/Sil-OH and 150 °C for Ir/Sil-F as well as Ir/Sil catalysts. For all catalysts, a further increase in temperature caused a decrease in the activity. The decrease of the catalytic activity at higher temperatures is explained in the literature by the occurrence of dehydrogenation [
35] or/and cracking of the product of the hydrogenation of toluene—methylcyclohexane [
36]. We did not observe any cracking products so the only possible explanation of the decrease in the catalytic activity at 150 °C was the dehydrogenation of methylcyclohexane, which was the only reaction product observed. The Wheeler-Weisz modulus (φ
2η) [
37] estimated for all investigated catalysts were lower than 1 within the temperature range used in this work, allowing to ignore diffusional limitations in applied experimental conditions and their influence on the activity of the investigated catalysts.
Our earlier studies indicated the weak Lewis acid sites on the surface of silicalite-1 modified with NH
4OH, whereas the modification with NH
4F caused the generation of strong Lewis acid sites [
16]. The presence of weak Lewis acid centers has a positive effect on the hydrogenation of toluene facilitating the adsorption and activation of the aromatic ring. It is in agreement with the findings of Lin et al. [
38] showing that the conversion of aromatics on the metal/acid catalysts takes place on two kinds of sites: metal active center and a neighboring acid site on which the adsorbed aromatics can be hydrogenated with the activated hydrogen spillover from metal centers.
Figure 9A presents a plot of catalytic activity as a function of the process temperature for the three reaction runs carried out on the most active catalyst, Ir/Sil-OH. The same catalyst was used in three cycles of measurements, with changing the temperature in the range of 75 ↗ 225 °C in cycle 1, 225 ↘ 100 °C in cycle 2, and 100 ↗ 200 °C in cycle 3. At each temperature two measurements were performed at 10 min intervals—the plot was made using the averaged values. As follows from the plot, only in the second cycle a small decrease in the activity was observed in the temperature range of 175–125 °C. This decrease did not exceed 10% of the initial activity, which means that the activity was maintained at a stable level. The catalyst activity in the third cycle was almost the same as that in the second cycle. A similar decrease in activity was observed for Ir/Sil-OH catalyst in the 22 h stability test (
Figure 9B). The most active catalysts (Ir/Sil-OH and Ir/Sil-F) were tested at 125 °C for a long period of time (22 h) to check their stability. In the evaluated period of time, the tested catalysts showed stable work. The activity of both catalysts increased for the first 30–40 min of the reaction to the level of ~16.5 and 11.5 mol
Tl⋅mol
Ir−1⋅min
−1, respectively, and then it gradually decreased and stabilized after a few hours. The activity of Ir/Sil-OH in the 22 h test decreased by less than 10%, while the activity of Ir/Sil-F fell by 15%.
A comparison of the TOF (48.9 min
−1) values obtained for the best Ir/Sil-OH catalyst with the results reported in the literature for iridium supported on different supports (SBA-3, Al
2O
3, SiO
2 [
39,
40], MgF
2 [
19], magnesium oxo-fluoride [
41,
42]) shows that the iridium catalyst supported on modified silicalite-1 is a more promising catalyst—
Table 6.
Based on the paper [
43], presenting the influence of different active phases (Pt, Ir, Ru) on catalytic activity for toluene hydrogenation, it can be concluded that iridium may have a great potential for the hydrogenation of aromatic compounds.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}