
Machine Learning Prediction of the Redox Activity of Quinones



Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset Construction
2.1.1. Molecular Structure Generation
2.1.2. Dataset Splitting
2.2. Molecular Descriptors
- Strings—for example, the SMILSES representation that contains information about atom types and connectivity [28];
- Connection table formats [29]: tabular formats that provide information about atom counts, atom types, connectivity matrix, bonded pairs of atoms, chirality, etc.; an example for such molecular representation format is the MDL molfile;
- Vectors of features: a molecule can be represented either as a vector of molecular properties (descriptors) such as molecular weight, molecular volume, numbers of certain atom types, topology, etc., [30] or as a molecular fingerprint: a bitstring (can be regarded as vector of ones and zeros) is derived from the molecular structure according to a predefined set of rules [31]—among the most employed fingerprints are the extended-connectivity fingerprints (ECFPs) based on Morgan’s algorithm [32], since they are specially designed for establishing structure–property relationships [33];
- Computer-learned representations: in recent years, a large number of machine learning-based molecular representations were developed—those methods rely on convolutional neural networks (CNNs) and/or recurrent neural networks (RNNs) to transform a molecule represented as a SMILES string or as 3D Cartesian atom coordinates to a low dimensional latent space [34,35] that can be used both for property prediction and for the generation of new molecular structures [36].
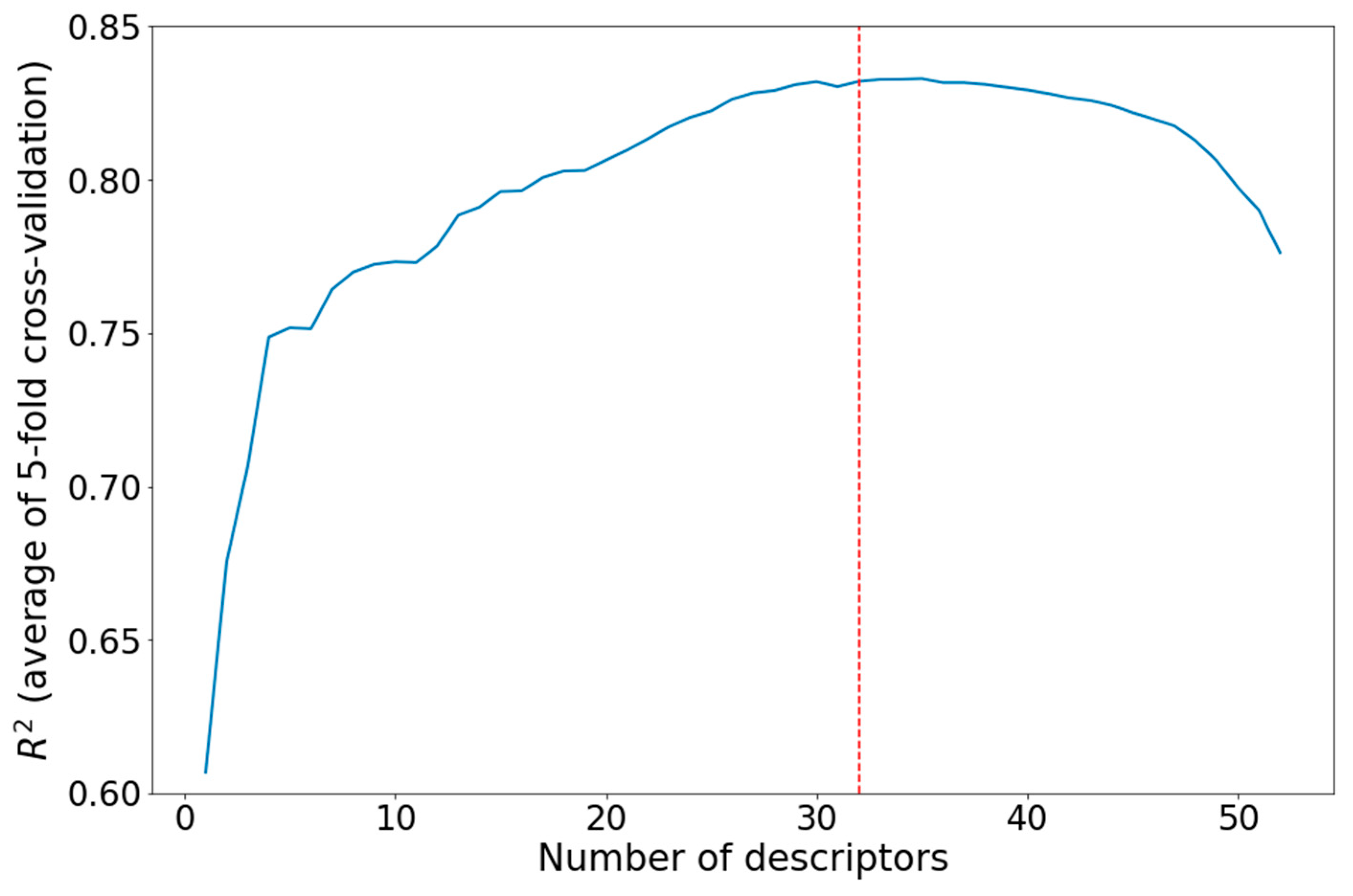
Descriptor Selection
- Low-variance descriptors were removed: descriptors whose value equalled the descriptor mode for 60% or more of the molecules in the dataset were discarded;
- Descriptors that had a weak correlation with the target value were discarded. Correlations with covariance between the normalized descriptors and normalized target values of less than 0.25 were considered as weak correlations. The normalization was performed as follows:where Vnorm is the normalized value, V is the unnormalized value, Vmean is the mean of V in the dataset, and σV is the standard deviation of V in the dataset.
- Strongly mutually correlated descriptors (covariance between normalized descriptors greater than 0.7) were discarded. After this operation, 52 descriptors were left;
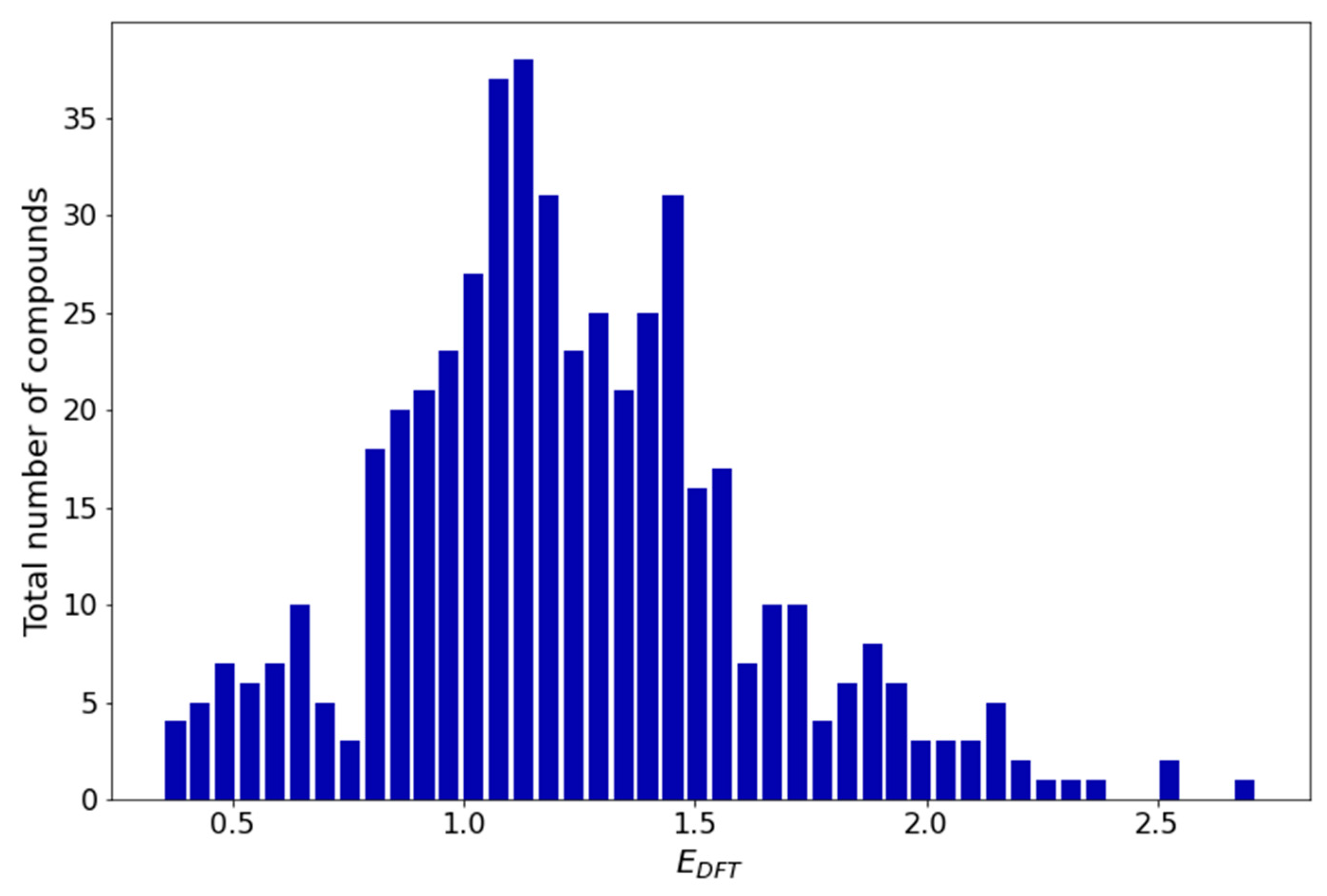
2.3. Redox Potential Calculation

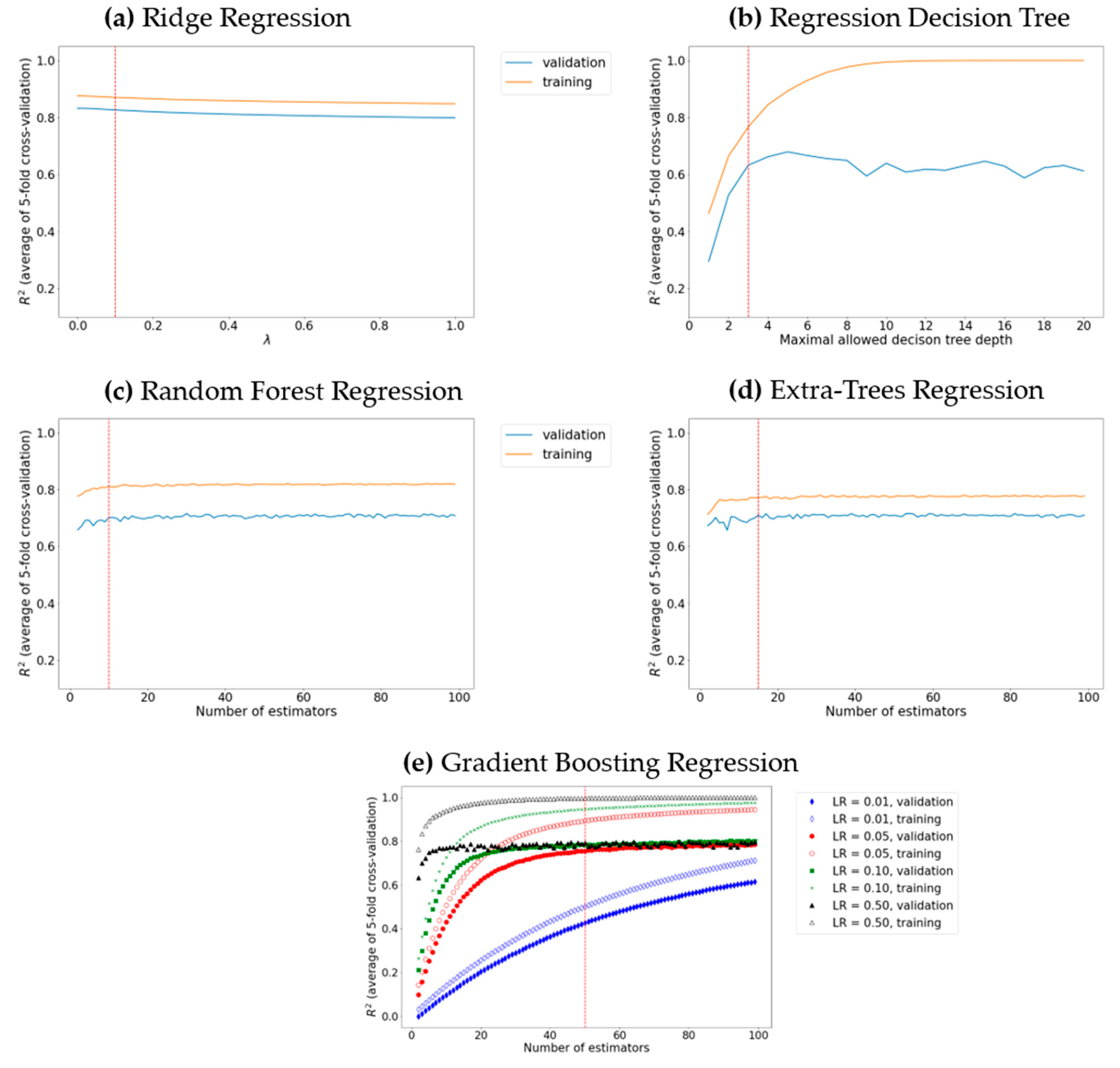
2.4. Machine Learning Methods Used
2.4.1. Ridge Regression
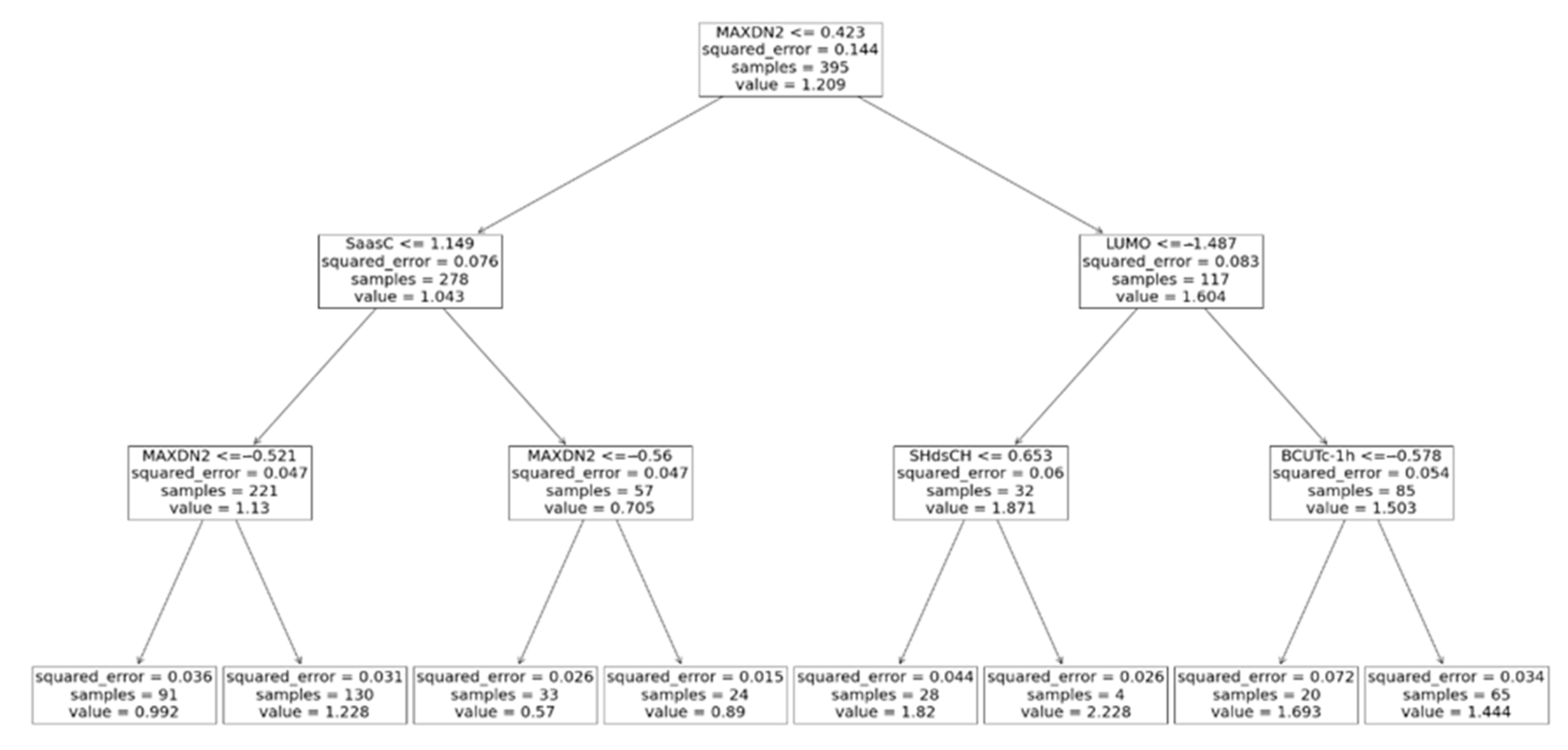
2.4.2. Decision Tree
2.4.3. Random Forest
2.4.4. Extra Trees
2.4.5. Gradient Boosting
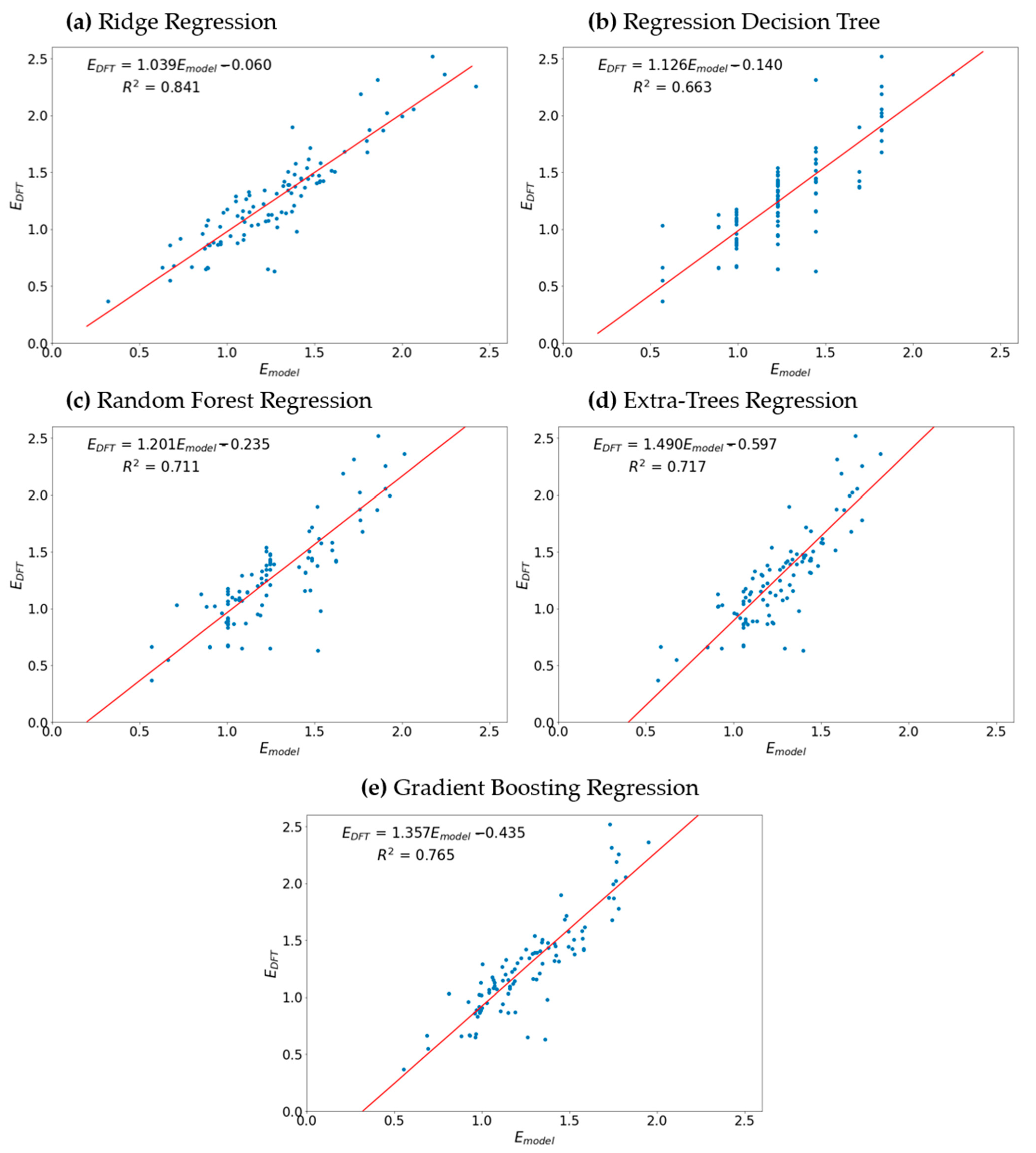
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poizot, P.; Dolhem, F. Clean energy new deal for a sustainable world: From non-CO2 generating energy sources to greener electrochemical storage devices. Energy Environ. Sci. 2011, 4, 2003–2019. [Google Scholar] [CrossRef]
- Larcher, D.; Tarascon, J.M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 2015, 7, 19–29. [Google Scholar] [CrossRef]
- Poizot, P.; Gaubicher, J.; Renault, S.; Dubois, L.; Liang, Y.; Yao, Y. Opportunities and Challenges for Organic Electrodes in Electrochemical Energy Storage. Chem. Rev. 2020, 120, 6490–6557. [Google Scholar] [CrossRef]
- Schon, T.B.; McAllister, B.T.; Li, P.-F.; Seferos, D.S. The rise of organic electrode materials for energy storage. Chem. Soc. Rev. 2016, 45, 6345–6404. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, Q.; Li, L.; Niu, Z.; Chen, J. Design Strategies toward Enhancing the Performance of Organic Electrode Materials in Metal-Ion Batteries. Chemistry 2018, 4, 2786–2813. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J. Prospects of organic electrode materials for practical lithium batteries. Nat. Rev. Chem. 2020, 4, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Esser, B.; Dolhem, F.; Becuwe, M.; Poizot, P.; Vlad, A.; Brandell, D. A perspective on organic electrode materials and technologies for next generation batteries. J. Power Sources 2021, 482, 228814. [Google Scholar] [CrossRef]
- Yan, L.; Zhao, C.; Sha, Y.; Li, Z.; Liu, T.; Ling, M.; Zhou, S.; Liang, C. Electrochemical redox behavior of organic quinone compounds in aqueous metal ion electrolytes. Nano Energy 2020, 73, 10476. [Google Scholar] [CrossRef]
- Tobishima, S.; Yamaki, J.; Yamaji, A. Cathode Characteristics of Organic Electron. Acceptors for Lithium Batteries. J. Electrochem. Soc. 1984, 131, 57–63. [Google Scholar] [CrossRef]
- Senoh, H.; Yao, M.; Sakaebe, H.; Yasuda, K.; Siroma, Z. A two-compartment cell for using soluble benzoquinone derivatives as active materials in lithium secondary batteries. Electrochim. Acta 2011, 56, 10145–10150. [Google Scholar] [CrossRef]
- Genorio, B.; Pirnat, K.; Cerc-Korosec, R.; Dominko, R.; Gaberscek, M. Electroactive Organic Molecules Immobilized onto Solid Nanoparticles as a Cathode Material for Lithium-Ion Batteries. Angew. Chem. Int. Ed. 2010, 49, 7222–7224. [Google Scholar] [CrossRef]
- Foos, J.S.; Erker, S.M.; Rembetsy, L.M. Synthesis and Characterization of Semiconductive Poly-l,4-Dirnethoxybenzene and Its Derived Polyquinone. J. Electrochem. Soc. 1986, 133, 836–840. [Google Scholar] [CrossRef]
- Häringer, D.; Novák, P.; Haas, O.; Piro, B.; Pham, M.-C. Poly(5-amino-1,4-naphthoquinone), a Novel Lithium-Inserting Electroactive Polymer with High Specific Charge. J. Electrochem. Soc. 1999, 146, 2393–2396. [Google Scholar] [CrossRef]
- Gall, T.L.; Reiman, K.H.; Grossel, M.C.; Owen, J.R. Poly(2,5-dihydroxy-1,4-benzoquinone-3,6-methylene): A new organic polymer as positive electrode material for rechargeable lithium batteries, Journal of Power Sources. J. Power Sources 2003, 119–121, 316–320. [Google Scholar] [CrossRef]
- Son, E.J.; Kim, J.H.; Kim, K.; Park, C.B. Quinone and its derivatives for energy harvesting and storage materials. J. Mat. Chem. A 2016, 4, 11179–11202. [Google Scholar] [CrossRef]
- Chambers, J.Q. Quinonoid Compounds, 1st ed.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Mueller, T.; Kusne, A.G.; Ramprasad, R. Machine learning in materials science: Recent progress and emerging applications. In Reviews in Computational Chemistry; Parrill, A.L., Lipkowitz, B.K., Eds.; John Wiley & Sons, Inc.: Indianapolis, IN, USA, 2016; Volume 29, pp. 186–273. [Google Scholar]
- Joshi, R.P.; Eickholt, L.; Li, L.; Fornari, M.; Barone, V.; Peralta, J.E. Machine Learning the Voltage of Electrode Materials in Metal-ion Batteries. ACS Appl. Mater. Interfaces 2019, 11, 18494–18503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, J.; Lu, J.; Shen, L. Interpretable learning of voltage for electrode design of multivalent metal-ion batteries. NPJ Comput. Mater. 2022, 8, 175. [Google Scholar] [CrossRef]
- Allam, O.; Kuramshin, R.; Stoichev, Z.; Cho, B.W.; Lee, S.W.; Jang, S.S. Molecular structure–redox potential relationship for organic electrode materials: Density functional theory–Machine learning approach. Mater. Today Energy 2020, 17, 100482. [Google Scholar] [CrossRef]
- Tuttle, M.R.; Brackman, E.M.; Sorourifar, F.; Paulson, J.; Zhang, S. Predicting the Solubility of Organic Energy Storage Materials Based on Functional Group Identity and Substitution Pattern. J. Phys. Chem. Lett. 2023, 14, 1318–1325. [Google Scholar] [CrossRef]
- Wang, F.; Li, J.; Liu, Z.; Qiu, T.; Wu, J.; Lu, D. Computational design of quinone electrolytes for redox flow batteries using high-throughput machine learning and theoretical calculations. Front. Chem. Eng. 2022, 4, 1086412. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Open Babel Development Team. Open Babel. 2016. Available online: http://openbabel.org/wiki/Main_Page (accessed on 12 October 2023).
- Kichev, I.; Borislavov, L.; Tadjer, A. Automated generation of molecular derivatives—DerGen software package. Mater. Today Proceedings 2022, 61, 1287–1291. [Google Scholar] [CrossRef]
- Nayak, S.K.; Ojha, A.C. Data Leakage Detection and Prevention: Review and Research Directions. In Machine Learning and Information Processing. Advances in Intelligent Systems and Computing; Swain, D., Pattnaik, P., Gupta, P., Eds.; Springer: Singapore, 2020; Volume 1101, pp. 203–212. [Google Scholar]
- Wigh, D.S.; Goodman, J.M.; Lapki, A.A. A review of molecular representation in the age of machine learning. WIREs Comput. Mol. Sci. 2022, 12, e1603. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Dalby, A.; Nourse, J.G.; Hounshell, W.G.; Gushurst, A.K.I.; Grier, D.L.; Leland, B.A.; Laufer, J. Description of several chemical structure file formats used by computer programs developed at Molecular Design Limited. J. Chem. Inf. Comput. Sci. 1992, 32, 244–255. [Google Scholar] [CrossRef]
- Todeschini, R. Consonni Handbook of Molecular Descriptors. In Methods and Principles in Medicinal Chemistry; WILEY-VCH Verlag GmbH: Weinheim, Germany, 2000. [Google Scholar]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.L. The Generation of a Unique Machine Description for Chemical Structures-A Technique Developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5, 107–113. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.D.; Chung, P.W. Deep learning for molecular design—A review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar] [CrossRef]
- Kuzminykh, D.; Polykovskiy, D.; Kadurin, A.; Zhebrak, A.; Baskov, I.; Nikolenko, S.; Shayakhmetov, R.; Zhavoronkov, A. 3D Molecular Representations Based on the Wave Transform for Convolutional Neural Networks. Mol. Pharm. 2018, 15, 4378–4385. [Google Scholar] [CrossRef] [PubMed]
- Skalic, M.; Jiménez Luna, J.; Sabbadin, D.; De Fabritiis, G. Shape-Based Generative Modeling for de-novo Drug Design. J. Chem. Inf. Model. 2019, 59, 1205–1214. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- James, J.P. Stewart, Stewart Computational Chemistry; MOPAC: Colorado Springs, CO, USA, 2016; Available online: http://OpenMOPAC.net (accessed on 12 October 2023).
- Pudjihartono, N.; Fadason, T.; Kempa-Liehr, A.W.; O’Sullivan, J.M. A Review of Feature Selection Methods for Machine Learning-Based Disease Risk Prediction. Front. Bioinform. 2022, 2, 927312. [Google Scholar] [CrossRef] [PubMed]
- Hocking, R.R. The Analysis and Selection of Variables in Linear Regression. Biometrics 1976, 32, 1–49. [Google Scholar] [CrossRef]
- Burden, F.R. Molecular identification number for substructure searches. J. Chem. Inf. Comput. Sci. 1989, 29, 225–227. [Google Scholar] [CrossRef]
- Sharma, V.; Goswami, R.; Madan, A.K. Eccentric Connectivity Index: A Novel Highly Discriminating Topological Descriptor for Structure-Property and Structure-Activity Studies. J. Chem. Inf. Comput. Sci. 1997, 37, 273–282. [Google Scholar] [CrossRef]
- Hall, L.H.; Kier, L.B. Electrotopological state indices for atom types: A novel combination of electronic, topological, and valence state information. J. Chem. Inf. Comput. Sci. 1995, 35, 1039–1045. [Google Scholar] [CrossRef]
- Gramatica, P.; Corradi, M.; Consonni, V. Modelling and prediction of soil sorption coefficients of non-ionic organic pesticides by molecular descriptors. Chemosphere 2000, 41, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Nilakantan, R.; Nunn, D.S.; Greenblatt, L.; Walker, G.; Haraki, K.; Mobilio, D. A family of ring system-based structural fragments for use in structure-activity studies: Database mining and recursive partitioning. J. Chem. Inf. Model. 2006, 46, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics; WILEY-VCH Verlag GmbH: Weinheim, Germany, 2009; pp. 809–812. [Google Scholar]
- Zhao, Y.H.; Abraham, M.H.; Zissimos, A.M. Fast Calculation of van der Waals Volume as a Sum of Atomic and Bond Contributions and Its Application to Drug Compounds. JACS 2003, 68, 7368–7373. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Ochterski, J.W. Thermochemistry in Gaussian; Gaussian, Inc.: Wallingford, CT, USA, 2000. [Google Scholar]
- Danchovski, Y.; Rasheev, H.; Stoyanova, R.; Tadjer, A. Molecular Engineering of Quinone-Based Nickel Complexes and Polymers for All-Organic Li-Ion Batteries. Molecules 2022, 27, 6805. [Google Scholar] [CrossRef] [PubMed]
- Hoerl, A.E.; Kennard, R.W. Ridge Regression: Biased Estimation for Nonorthogonal Problems. Technometrics 1970, 12, 55–67. [Google Scholar] [CrossRef]
- Breiman, L. Classification and Regression Trees, 1st ed.; CHAPMAN & HALL/CRC: Boca Raton, FL, USA, 1984. [Google Scholar]
- Kotsiantis, S.B. Decision trees: A recent overview. Artif. Intell. Rev. 2013, 39, 261–283. [Google Scholar] [CrossRef]
- Klekota, J.; Roth, F.P. Chemical substructures that enrich for biological activity. Bioinformatics 2008, 24, 2518–2525. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y. ADME evaluation in drug discovery. The prediction of human intestinal absorption by a support vector machine. J. Chem. Inf. Model. 2007, 47, 2408–2415. [Google Scholar] [CrossRef] [PubMed]
- Lamanna, C.; Bellini, M.; Padova, A.; Westerberg, G.; Maccari, L. Straightforward recursive partitioning model for discarding insoluble compounds in the drug discovery process. J. Med. Chem. 2008, 51, 2891–2897. [Google Scholar] [CrossRef]
- Patel, H.H.; Prajapati, P. Study and Analysis of Decision Tree Based Classification Algorithms. Int. J. Comput. Sci. Eng. 2018, 6, 74–78. [Google Scholar] [CrossRef]
- Ho, T.K. The random subspace method for constructing decision forests. IEEE Trans. Pattern Anal. Mach. Intell. 1998, 20, 832–844. [Google Scholar]
- Geurts, P.; Ernst, D.; Wehenkel, L. Extremely randomized trees. Mach. Learn. 2006, 63, 3–42. [Google Scholar] [CrossRef]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Piryonesi, S.M.; El-Diraby, T.E. Data Analytics in Asset Management: Cost-Effective Prediction of the Pavement Condition Index. J. Infrastruct. Syst. 2020, 26, 04019036. [Google Scholar] [CrossRef]
- Pedregosa, D.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Bejani, M.M.; Ghatee, M. A systematic review on overfitting control in shallow and deep neural networks. Artif. Intell. Rev. 2021, 54, 6391–6438. [Google Scholar] [CrossRef]
- Wright, S. Correlation and causation. J. Agric. Res. 1921, 20, 557–585. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Descriptor Name |
|---|---|
| Lowest partial charge weighted BCUTS [41] | BCUTc-1l |
| Highest partial charge weighted BCUTS [41] | BCUTc-1h |
| Total number of double bonds (excluding aromatic bonds) | nBondsD2 |
| Triply bound carbon bound to another carbon | C1SP1 |
| Doubly bound carbon bound to three other carbons | C3SP2 |
| A topological descriptor combining distance and adjacency information [42] | ECCEN |
| Count of atom-type H E-State: H on aaCH, dCH2 or dsCH * [43] | nHother |
| Count of atom-type E-State: =C< [43] | ndssC |
| Count of atom-type E-State: aaC- [43] | naasC |
| Count of atom-type E-State: N≡ [43] | ntN |
| Sum of E-States for weak hydrogen bond acceptors [43] | SwHBa |
| Sum of atom-type H E-State: =CH- [43] | SHdsCH |
| Sum of atom-type H E-State: H on aaCH, dCH2 or dsCH [43] | SHother |
| Sum of atom-type E-State: =C< [43] | SdssC |
| Sum of atom-type E-State: aaC- [43] | SaasC |
| Sum of atom-type E-State: N≡ [43] | StN |
| Minimum atom-type H E-State: H on aaCH, dCH2 or dsCH [43] | minHother |
| Minimum atom-type E-State: aaC- [43] | minaasC |
| Minimum atom-type E-State: =O [43] | mindO |
| Maximum atom-type H E-State: H on aaCH, dCH2 or dsCH [43] | maxHother |
| Maximum atom-type E-State: aaC- [43] | maxaasC |
| Mean intrinsic state values I [43] | meanI |
| Maximum negative intrinsic state difference in the molecule (related to the nucleophilicity of the molecule) [44] | MAXDN2 |
| Maximum positive intrinsic state difference in the molecule (related to the electrophilicity of the molecule) [44] | MAXDP2 |
| Complexity of the system [45] | fragC |
| Number of rings | nRing |
| Topological diameter (maximum atom eccentricity) | topoDiameter |
| Mean topological charge index of order two [46] | JGI2 |
| Topological polar surface area | TopoPSA |
| Van der Waals volume calculated using the method proposed in Zhao et al. JACS 2003, 68, 7368–7373 [47] | VABC |
| Molecular weight | MW |
| Energy of the lowest unoccupied molecular orbital estimated by PM6 [eV] | LUMO |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kichev, I.; Borislavov, L.; Tadjer, A.; Stoyanova, R. Machine Learning Prediction of the Redox Activity of Quinones. Materials 2023, 16, 6687. https://doi.org/10.3390/ma16206687
Kichev I, Borislavov L, Tadjer A, Stoyanova R. Machine Learning Prediction of the Redox Activity of Quinones. Materials. 2023; 16(20):6687. https://doi.org/10.3390/ma16206687
Chicago/Turabian StyleKichev, Ilia, Lyuben Borislavov, Alia Tadjer, and Radostina Stoyanova. 2023. "Machine Learning Prediction of the Redox Activity of Quinones" Materials 16, no. 20: 6687. https://doi.org/10.3390/ma16206687
APA StyleKichev, I., Borislavov, L., Tadjer, A., & Stoyanova, R. (2023). Machine Learning Prediction of the Redox Activity of Quinones. Materials, 16(20), 6687. https://doi.org/10.3390/ma16206687