E3L and F1L Gene Functions Modulate the Protective Capacity of Modified Vaccinia Virus Ankara Immunization in Murine Model of Human Smallpox

Abstract
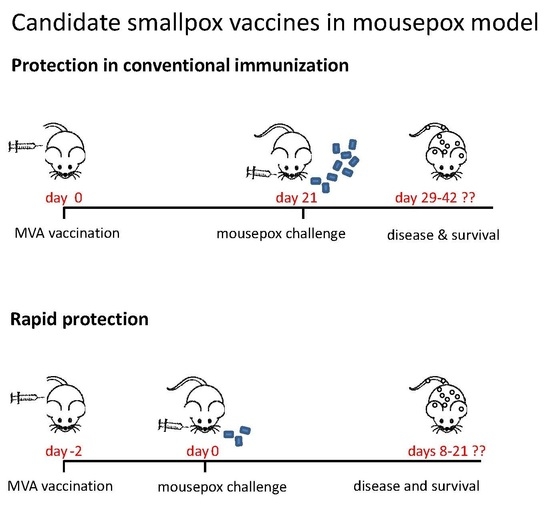
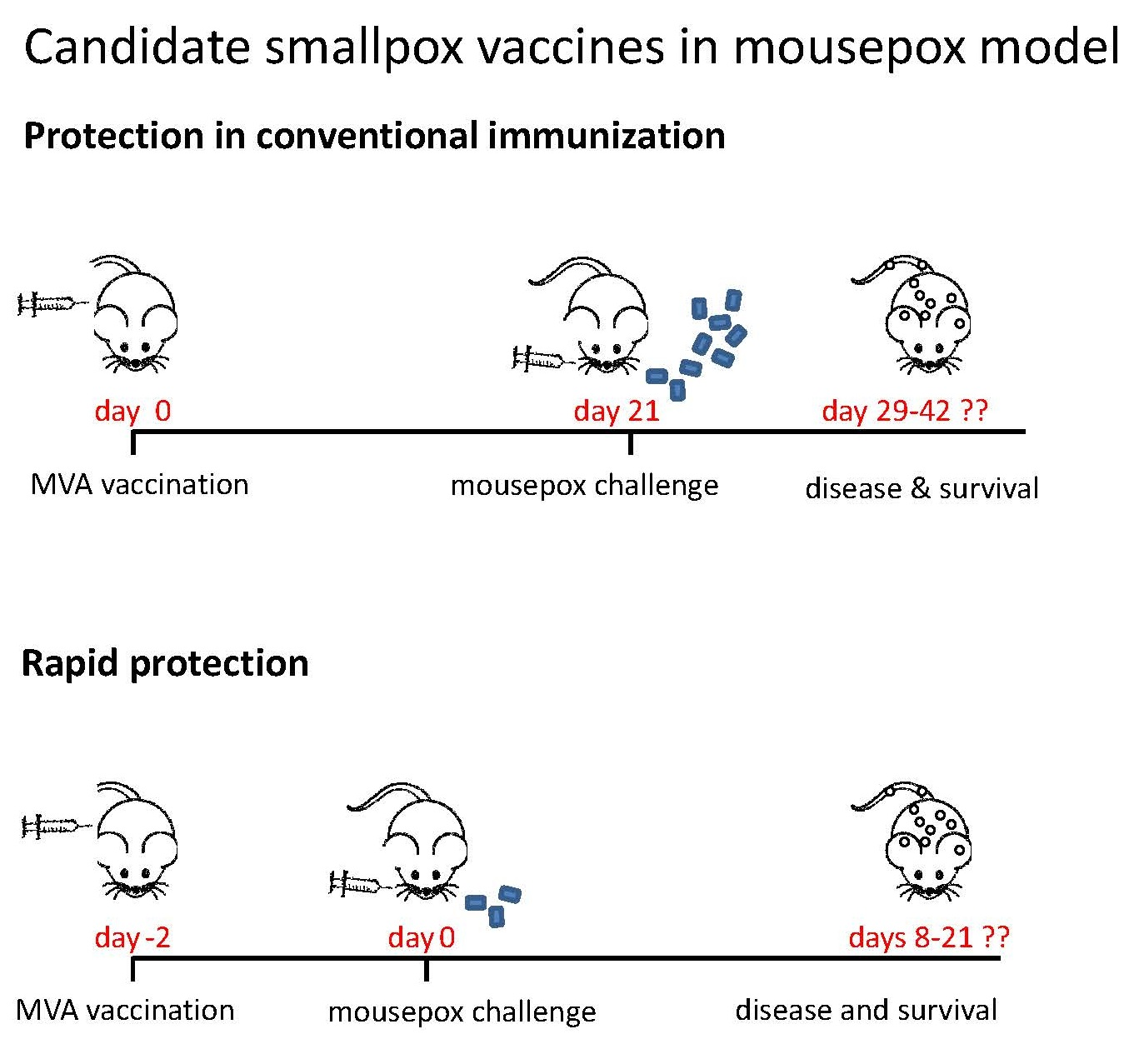
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Western Blot Analysis
2.3. Northern Blot Analysis
2.4. Mice
2.5. Immunization and Infection Experiments
2.6. Analysis of MVA-Specific Antibodies
2.7. Analysis of MVA Neutralizing Antibodies
2.8. Analysis of Antigen-Specific CD8+ T Cells by Enzyme-Linked Immunospot Assay (ELISPOT)
2.9. Statistical Analysis
3. Results
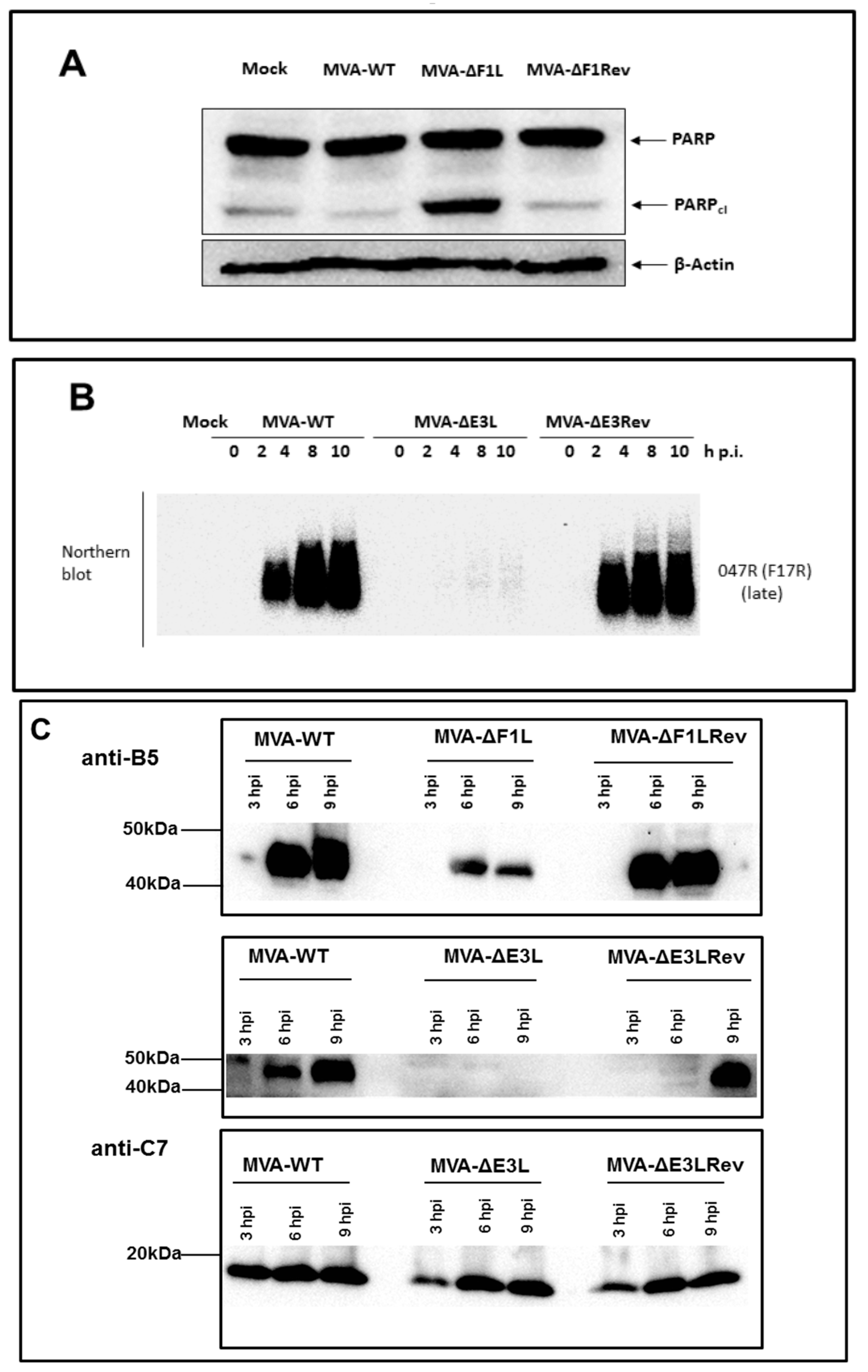
3.1. Decreased Synthesis of the Late Viral Protein B5 in the Absence of E3L or F1L Gene Expression
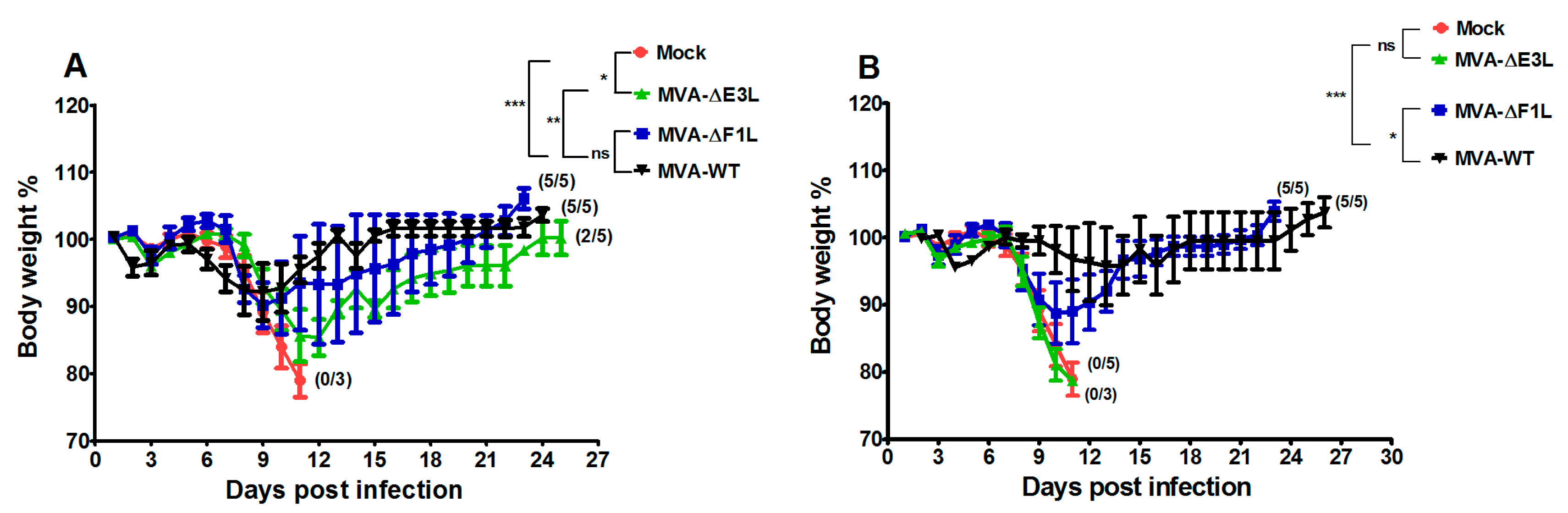
3.2. Protective Capacity of MVA Deletion Viruses against Lethal Poxvirus Infections
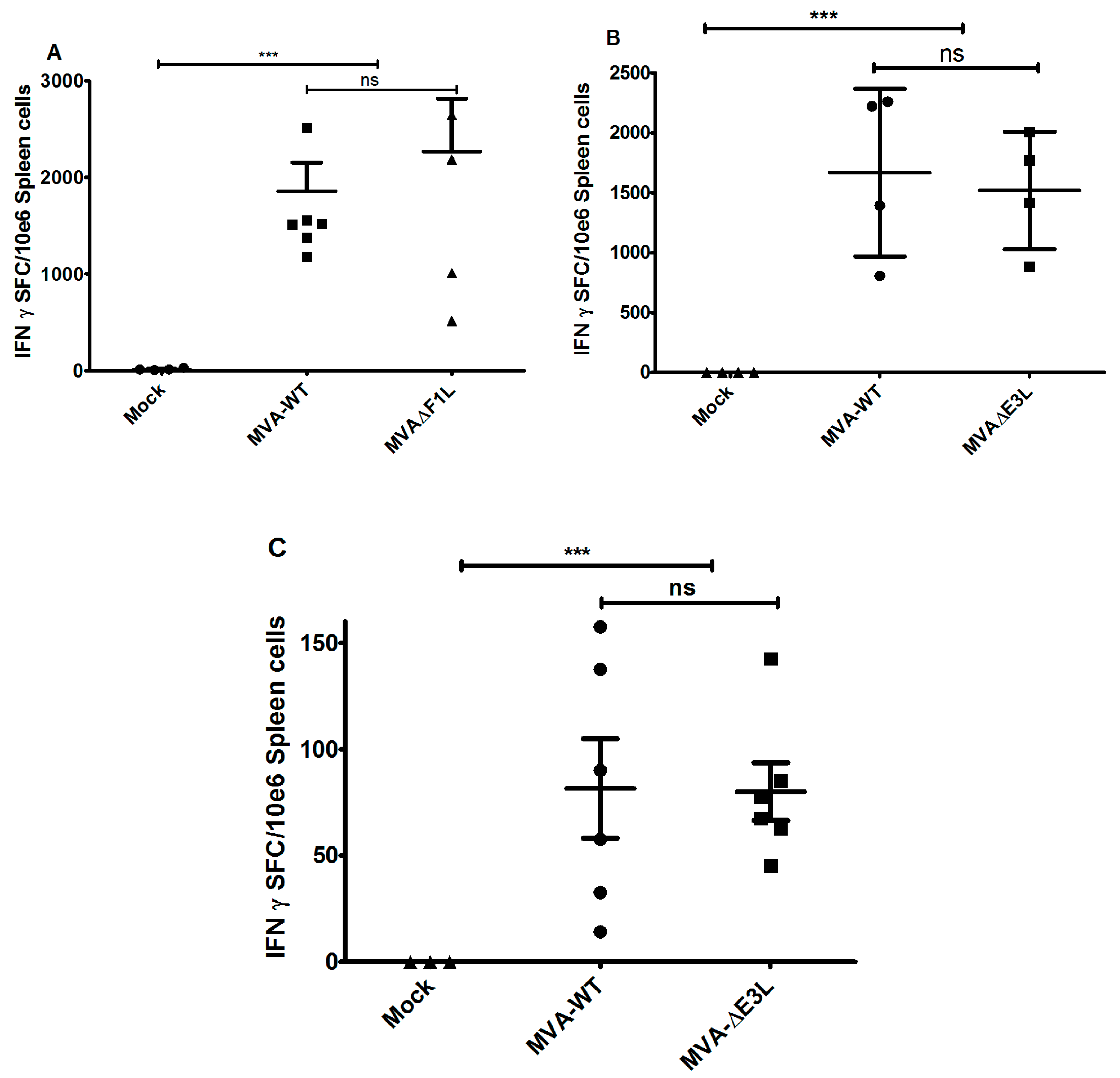
3.3. MVA-ΔF1L or MVA-ΔE3L Vaccines Prime Unimpaired VACV-Specific CD8+ T-Cell Responses
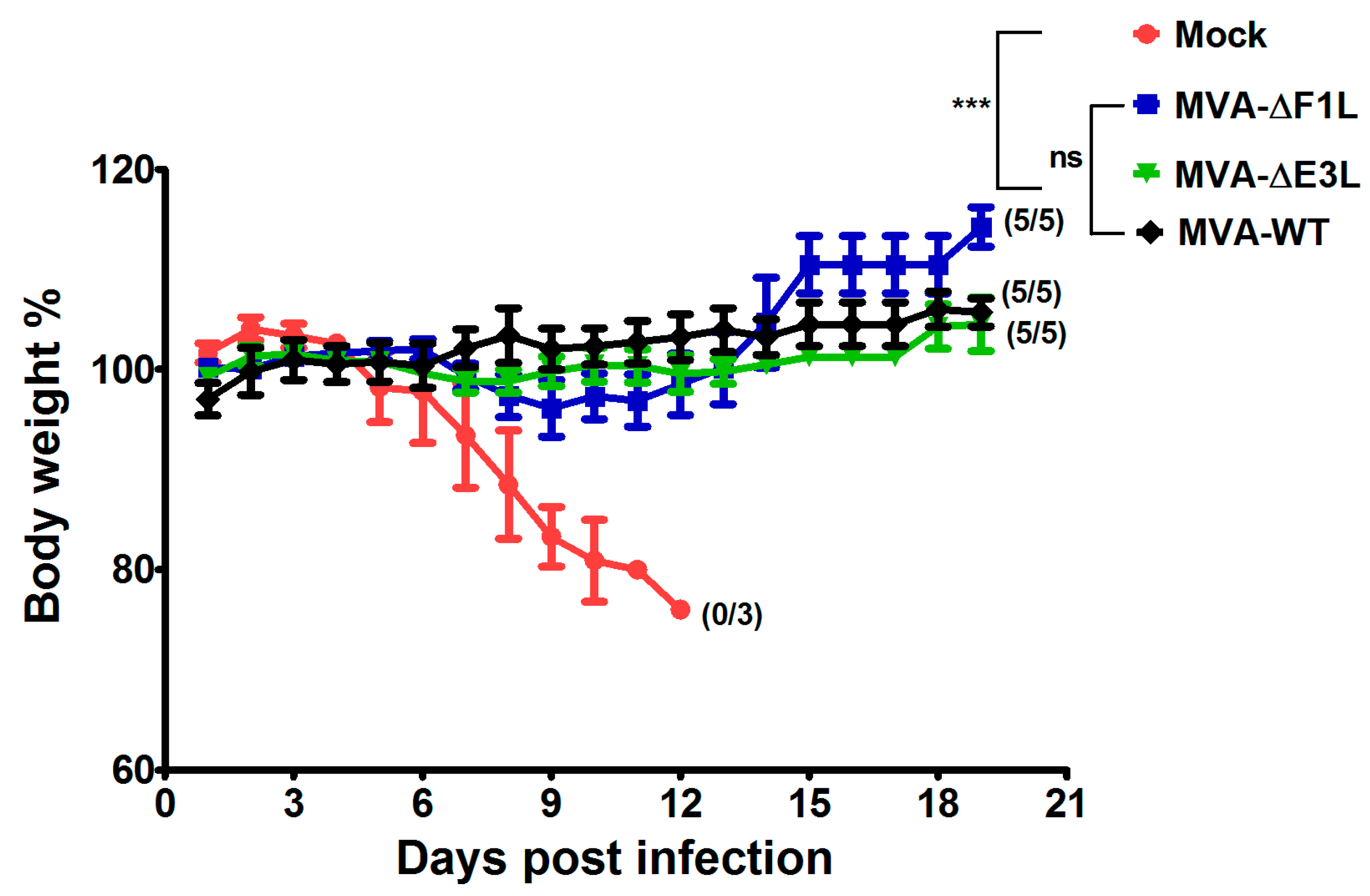
3.4. Rapidly Protective Immunization with MVA Vaccines Deficient in E3L or F1L
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Volz, A.; Sutter, G. Protective efficacy of modified vaccinia virus Ankara in preclinical studies. Vaccine 2013, 31, 4235–4240. [Google Scholar] [CrossRef] [PubMed]
- Volz, A.; Sutter, G. Chapter five—Modified vaccinia virus Ankara: History, value in basic research, and current perspectives for vaccine development. In Advances in Virus Research; Kielian, M., Mettenleiter, T.C., Roossinck, M.J., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 97, pp. 187–243. [Google Scholar]
- Volz, A.; Langenmayer, M.; Jany, S.; Kalinke, U.; Sutter, G. Rapid expansion of CD8+ T cells in wild-type and type I interferon receptor-deficient mice correlates with protection after low-dose emergency immunization with modified vaccinia virus Ankara. J. Virol. 2014, 88, 10946–10957. [Google Scholar] [CrossRef] [PubMed]
- Kremer, M.; Suezer, Y.; Volz, A.; Frenz, T.; Majzoub, M.; Hanschmann, K.-M.; Lehmann, M.H.; Kalinke, U.; Sutter, G. Critical role of perforin-dependent CD8+ T cell immunity for rapid protective vaccination in a murine model for human smallpox. PLoS Pathog. 2012, 8, e1002557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paran, N.; Suezer, Y.; Lustig, S.; Israely, T.; Schwantes, A.; Melamed, S.; Katz, L.; Preuß, T.; Hanschmann, K.-M.; Kalinke, U.; et al. Postexposure immunization with modified vaccinia virus Ankara or conventional lister vaccine provides solid protection in a murine model of human smallpox. J. Infect. Dis. 2009, 199, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.H.; Kenyon, J.C.; Bartlett, N.W.; Tscharke, D.C.; Smith, G.L. Deletion of gene A41L enhances vaccinia virus immunogenicity and vaccine efficacy. J. Gener. Virol. 2006, 87, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Staib, C.; Kisling, S.; Erfle, V.; Sutter, G. Inactivation of the viral interleukin 1β receptor improves CD8+ T-cell memory responses elicited upon immunization with modified vaccinia virus Ankara. J. Gen. Virol. 2005, 86, 1997–2006. [Google Scholar] [CrossRef] [PubMed]
- Banadyga, L.; Gerig, J.; Stewart, T.; Barry, M. Fowlpox virus encodes a bcl-2 homologue that protects cells from apoptotic death through interaction with the proapoptotic protein bak. J. Virol. 2007, 81, 11032–11045. [Google Scholar] [CrossRef] [PubMed]
- Cooray, S.; Bahar, M.W.; Abrescia, N.G.A.; McVey, C.E.; Bartlett, N.W.; Chen, R.A.-J.; Stuart, D.I.; Grimes, J.M.; Smith, G.L. Functional and structural studies of the vaccinia virus virulence factor N1 reveal a bcl-2-like anti-apoptotic protein. J. Gen. Virol. 2007, 88, 1656–1666. [Google Scholar] [CrossRef] [PubMed]
- Gubser, C.; Bergamaschi, D.; Hollinshead, M.; Lu, X.; van Kuppeveld, F.J.M.; Smith, G.L. A new inhibitor of apoptosis from vaccinia virus and eukaryotes. PLOS Pathog. 2007, 3, e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.M.; Quilty, D.; Banadyga, L.; Barry, M. The vaccinia virus protein F1L interacts with bim and inhibits activation of the pro-apoptotic protein bax. J. Biol. Chem. 2006, 281, 39728–39739. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, G.; Kastenmuller, W.; Ljapoci, R.; Sutter, G.; Drexler, I. Cross-priming of cytotoxic T cells dictates antigen requisites for modified vaccinia virus Ankara vector vaccines. J. Virol. 2007, 81, 11925–11936. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Vandenberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Van Gool, S.W.; Agostinis, P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci. Transl. Med. 2016, 8, 328ra327. [Google Scholar] [CrossRef] [PubMed]
- Behar, S.M.; Martin, C.J.; Booty, M.G.; Nishimura, T.; Zhao, X.; Gan, H.X.; Divangahi, M.; Remold, H.G. Apoptosis is an innate defense function of macrophages against mycobacterium tuberculosis. Mucosal Immunol. 2011, 4, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Fujii, T.; Ishijima, R.; Tachibana, H.; Yokoue, N.; Takasawa, R.; Tanuma, S.-I. The release of high mobility group box 1 in apoptosis is triggered by nucleosomal DNA fragmentation. Arch. Biochem. Biophys. 2011, 506, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zapata, J.; Pauza, C.; Salvato, M. Modulation of SIV and HIV DNA vaccine immunity by fas-fasl signaling. Viruses 2015, 7, 1429–1453. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.-S.; Vasilevskaya, I.A.; Wang, S.-C.; Bair, C.-H.; Chang, W. Apoptosis and host restriction of vaccinia virus in RK13 cells. Virus Res. 1997, 52, 121–132. [Google Scholar] [CrossRef]
- Fischer, S.F.; Ludwig, H.; Holzapfel, J.; Kvansakul, M.; Chen, L.; Huang, D.C.S.; Sutter, G.; Knese, M.; Hacker, G. Modified vaccinia virus Ankara protein F1L is a novel BH3-domain-binding protein and acts together with the early viral protein E3L to block virus-associated apoptosis. Cell Death Differ. 2005, 13, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Hornemann, S.; Harlin, O.; Staib, C.; Kisling, S.; Erfle, V.; Kaspers, B.; Häcker, G.; Sutter, G. Replication of modified vaccinia virus Ankara in primary chicken embryo fibroblasts requires expression of the interferon resistance gene E3L. J. Virol. 2003, 77, 8394–8407. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, H.; Mages, J.; Staib, C.; Lehmann, M.H.; Lang, R.; Sutter, G. Role of viral factor E3L in modified vaccinia virus Ankara infection of human HeLa cells: Regulation of the virus life cycle and identification of differentially expressed host genes. J. Virol. 2005, 79, 2584–2596. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, H.; Suezer, Y.; Waibler, Z.; Kalinke, U.; Schnierle, B.S.; Sutter, G. Double-stranded RNA-binding protein E3 controls translation of viral intermediate RNA, marking an essential step in the life cycle of modified vaccinia virus Ankara. J. Gen. Virol. 2006, 87, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Wasilenko, S.T.; Banadyga, L.; Bond, D.; Barry, M. The vaccinia virus F1L protein interacts with the proapoptotic protein bak and inhibits bak activation. J. Virol. 2005, 79, 14031–14043. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. Bcl-2-regulated apoptosis: Mechanism and therapeutic potential. Curr. Opin. Immunol. 2007, 19, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Sutter, G.; Mayr, A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J. Gen. Virol. 1991, 72, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Kremer, M.; Volz, A.; Kreijtz, J.H.C.M.; Fux, R.; Lehmann, M.; Sutter, G. Easy and efficient protocols for working with recombinant vaccinia virus MVA. In Vaccinia Virus and Poxvirology; Isaacs, S.N., Ed.; Humana Press: New York, NY, USA, 2012; Volume 890, pp. 59–92. [Google Scholar]
- Tscharke, D.C.; Karupiah, G.; Zhou, J.; Palmore, T.; Irvine, K.R.; Haeryfar, S.M.M.; Williams, S.; Sidney, J.; Sette, A.; Bennink, J.R.; et al. Identification of poxvirus CD8+ T cell determinants to enable rational design and characterization of smallpox vaccines. J. Exp. Med. 2005, 201, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Tscharke, D.C.; Woo, W.-P.; Sakala, I.G.; Sidney, J.; Sette, A.; Moss, D.J.; Bennink, J.R.; Karupiah, G.; Yewdell, J.W. Poxvirus CD8+ T-cell determinants and cross-reactivity in BALB/c mice. J. Virol. 2006, 80, 6318–6323. [Google Scholar] [CrossRef] [PubMed]
- Putz, M.M.; Midgley, C.M.; Law, M.; Smith, G.L. Quantification of antibody responses against multiple antigens of the two infectious forms of vaccinia virus provides a benchmark for smallpox vaccination. Nat. Med. 2006, 12, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, S.; Brühl, P.; Mayrhofer, J.; Schmid, K.; Gerencer, M.; Falkner, F.G. The nonreplicating smallpox candidate vaccines defective vaccinia lister (dvv-l) and modified vaccinia Ankara (MVA) elicit robust long-term protection. Virology 2005, 341, 91–101. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency (EMEA). Imvanex: Modifiziertes Vacciniavirus Ankara, Lebend; EMA/490157/2013; European Medicines Agency: London, UK, 2013. [Google Scholar]
- Dai, P.; Wang, W.; Cao, H.; Avogadri, F.; Dai, L.; Drexler, I. Modified vaccinia virus Ankara triggers type I IFN production in murine conventional dendritic cells via a cGas/Sting-mediated cytosolic DNA-sensing pathway. PLoS Pathog. 2014, 10, e1003989. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.J.; Coban, C.; Kato, H.; Takahashi, K.; Torii, Y.; Takeshita, F.; Ludwig, H.; Sutter, G.; Suzuki, K.; Hemmi, H.; et al. A toll-like receptor–independent antiviral response induced by double-stranded b-form DNA. Nat. Immunol. 2005, 7, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.W.; Watson, J.C.; Jacobs, B.L. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1992, 89, 4825–4829. [Google Scholar] [CrossRef] [PubMed]
- Rivas, C.; Gil, J.; Mělková, Z.; Esteban, M.; Díaz-Guerra, M. Vaccinia virus e3l protein is an inhibitor of the interferon (IFN)-induced 2-5a synthetase enzyme. Virology 1998, 243, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Wolferstätter, M.; Schweneker, M.; Späth, M.; Lukassen, S.; Klingenberg, M.; Brinkmann, K.; Wielert, U.; Lauterbach, H.; Hochrein, H.; Chaplin, P.; et al. Recombinant modified vaccinia virus Ankara generating excess early double-stranded RNA transiently activates protein kinase R and triggers enhanced innate immune responses. J. Virol. 2014, 88, 14396–14411. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, B.; Gómez, C.E.; Nájera, J.L.; Sorzano, C.O.S.; Delaloye, J.; González-Sanz, R.; Jiménez, V.; Roger, T.; Calandra, T.; Pantaleo, G.; et al. Deletion of the viral anti-apoptotic gene F1L in the HIV/AIDS vaccine candidate MVA-C enhances immune responses against HIV-1 antigens. PLoS ONE 2012, 7, e48524. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, R.N.; Hay, C.M.; Stapleton, J.T.; Marbury, T.C.; Wagner, E.; Kreitmeir, E.; Röesch, S.; von Krempelhuber, A.; Young, P.; Nichols, R.; et al. A randomized, double-blind, placebo-controlled phase II trial investigating the safety and immunogenicity of modified vaccinia Ankara smallpox vaccine (mva-bn®) in 56–80-year-old subjects. PLoS ONE 2016, 11, e0157335. [Google Scholar] [CrossRef] [PubMed]
- Kreijtz, J.; Goeijenbier, M.; Moesker, F.M.; van den Dries, L.; Goeijenbier, S.; De Gruyter, H.M.; Lehmann, M.H.; Mutsert, G.; van de Vijver, D.; Volz, A.; et al. Safety and immunogenicity of a modified-vaccinia-virus-Ankara-based influenza a H5N1 vaccine: A randomised, double-blind phase 1/2a clinical trial. Lancet Infect. Dis. 2014, 14, 1196–1207. [Google Scholar] [CrossRef]
- Edghill-Smith, Y.; Golding, H.; Manischewitz, J.; King, L.R.; Scott, D.; Bray, M.; Nalca, A.; Hooper, J.W.; Whitehouse, C.A.; Schmitz, J.E.; et al. Smallpox vaccine-induced antibodies are necessary and sufficient for protection against monkeypox virus. Nat. Med. 2005, 11, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Fogg, C.; Lustig, S.; Whitbeck, J.C.; Eisenberg, R.J.; Cohen, G.H.; Moss, B. Protective immunity to vaccinia virus induced by vaccination with multiple recombinant outer membrane proteins of intracellular and extracellular virions. J. Virol. 2004, 78, 10230–10237. [Google Scholar] [CrossRef] [PubMed]
- Lustig, S.; Fogg, C.; Whitbeck, J.C.; Eisenberg, R.J.; Cohen, G.H.; Moss, B. Combinations of polyclonal or monoclonal antibodies to proteins of the outer membranes of the two infectious forms of vaccinia virus protect mice against a lethal respiratory challenge. J. Virol. 2005, 79, 13454–13462. [Google Scholar] [CrossRef] [PubMed]
- Gilchuk, I.; Gilchuk, P.; Sapparapu, G.; Lampley, R.; Singh, V.; Kose, N.; Blum, D.L.; Hughes, L.J.; Satheshkumar, P.S.; Townsend, M.B.; et al. Cross-neutralizing and protective human antibody specificities to poxvirus infections. Cell 2016, 167, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Smallpox vaccines: Targets of protective immunity. Immunol. Rev. 2011, 239, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Moutaftsi, M.; Peters, B.; Pasquetto, V.; Tscharke, D.C.; Sidney, J.; Bui, H.-H.; Grey, H.; Sette, A. A consensus epitope prediction approach identifies the breadth of murine TCD8+-cell responses to vaccinia virus. Nat. Biotechnol. 2006, 24, 817–819. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volz, A.; Jany, S.; Freudenstein, A.; Lantermann, M.; Ludwig, H.; Sutter, G. E3L and F1L Gene Functions Modulate the Protective Capacity of Modified Vaccinia Virus Ankara Immunization in Murine Model of Human Smallpox. Viruses 2018, 10, 21. https://doi.org/10.3390/v10010021
Volz A, Jany S, Freudenstein A, Lantermann M, Ludwig H, Sutter G. E3L and F1L Gene Functions Modulate the Protective Capacity of Modified Vaccinia Virus Ankara Immunization in Murine Model of Human Smallpox. Viruses. 2018; 10(1):21. https://doi.org/10.3390/v10010021
Chicago/Turabian StyleVolz, Asisa, Sylvia Jany, Astrid Freudenstein, Markus Lantermann, Holger Ludwig, and Gerd Sutter. 2018. "E3L and F1L Gene Functions Modulate the Protective Capacity of Modified Vaccinia Virus Ankara Immunization in Murine Model of Human Smallpox" Viruses 10, no. 1: 21. https://doi.org/10.3390/v10010021
APA StyleVolz, A., Jany, S., Freudenstein, A., Lantermann, M., Ludwig, H., & Sutter, G. (2018). E3L and F1L Gene Functions Modulate the Protective Capacity of Modified Vaccinia Virus Ankara Immunization in Murine Model of Human Smallpox. Viruses, 10(1), 21. https://doi.org/10.3390/v10010021