Evolution and Genetic Diversity of Porcine Circovirus 3 in China


Abstract
:1. Introduction
2. Material and Methods
2.1. Tissue Samples, DNA Extraction, PCR Amplification, and Sequencing
2.2. Sequence Collection, Alignment, and Phylogenetic Analysis
2.3. Selection Model Analysis
3. Results
3.1. Characterization of PCV3 Strains Circulating in Fujian
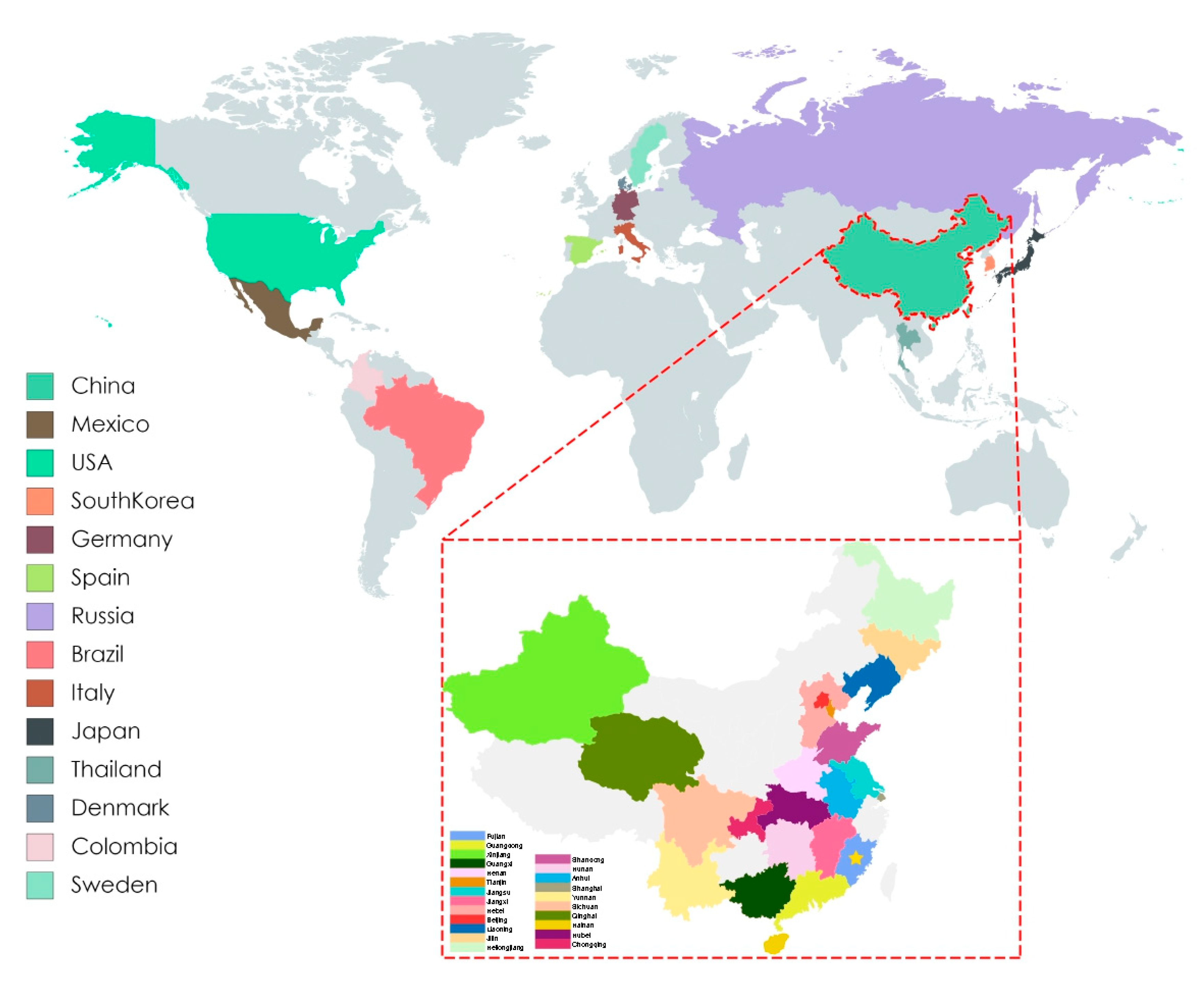
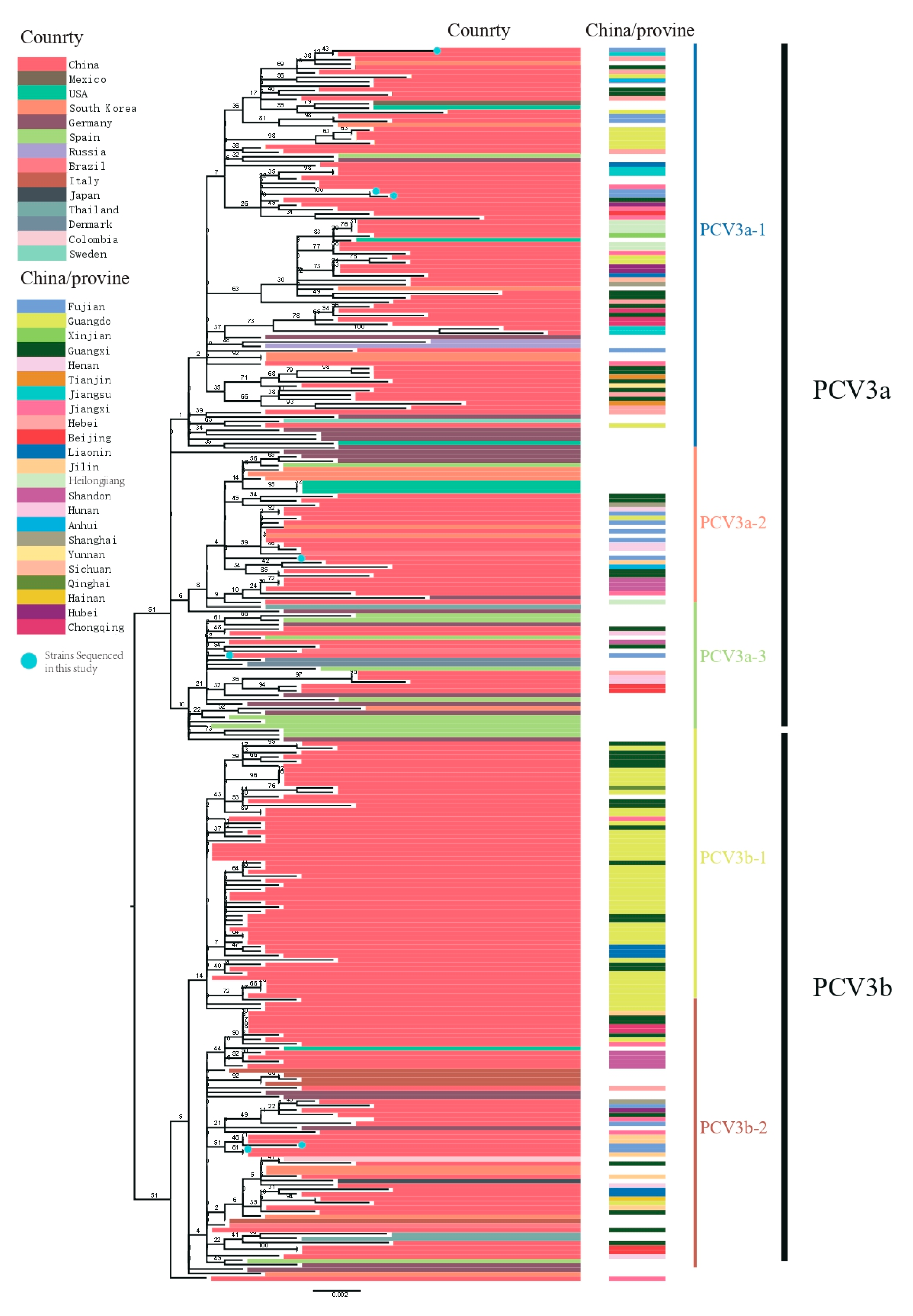
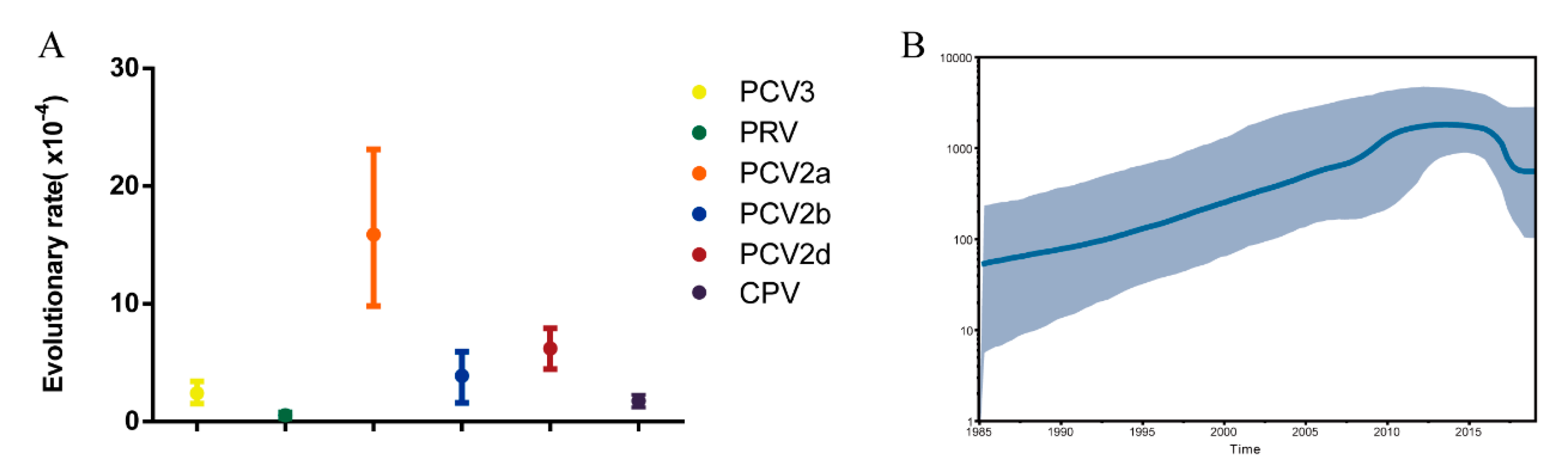
3.2. Phylogenetic and Evolution Analysis of PCV3 in China and Worldwide
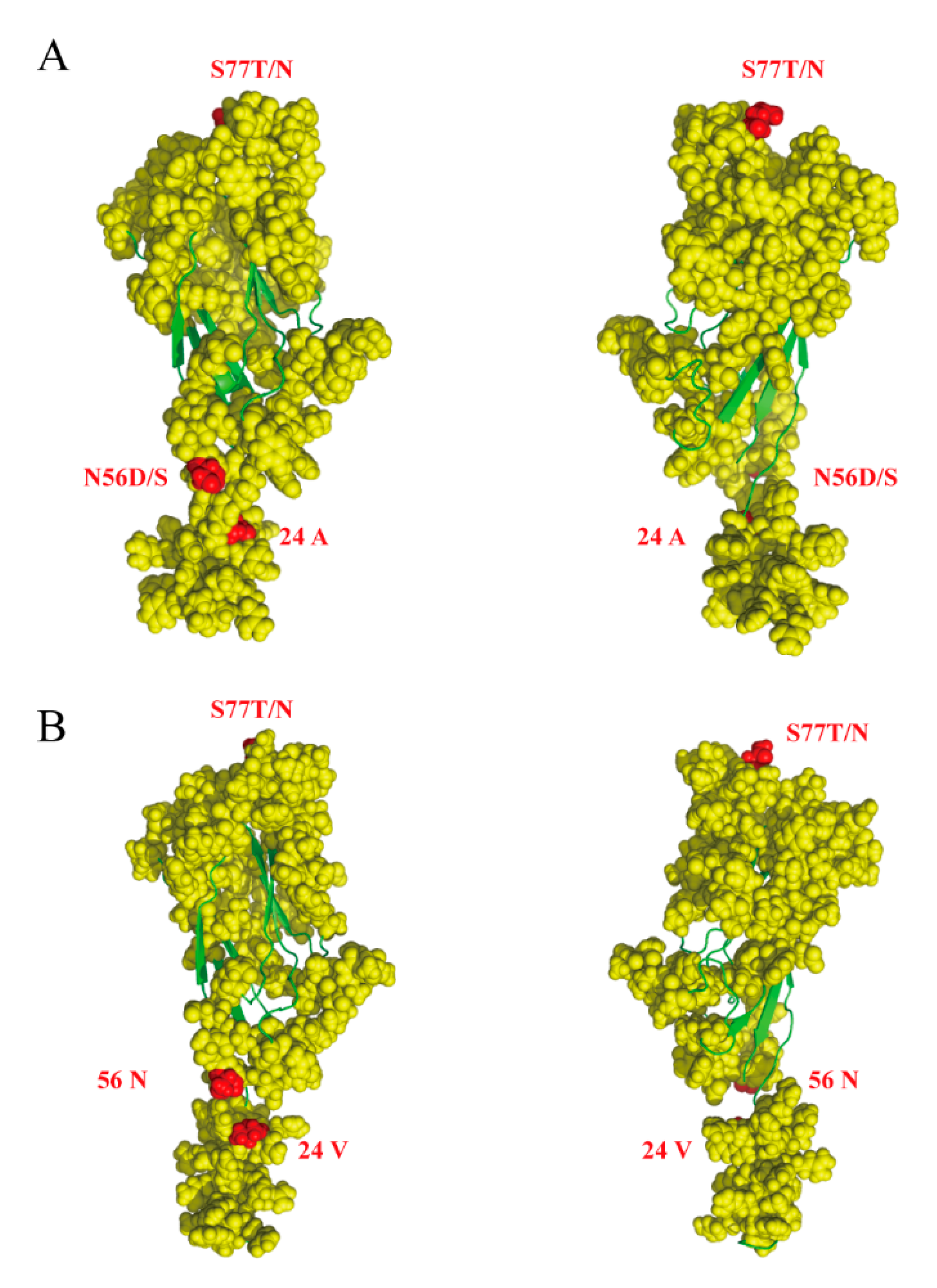
3.3. Selection and Amino Acid Function Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tischer, I.; Gelderblom, H.; Vettermann, W.; Koch, M.A. A very small porcine virus with circular single-stranded DNA. Nature 1982, 295, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Todd, D. Avian circovirus diseases: Lessons for the study of PMWS. Vet. Microbiol. 2004, 98, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Firth, C.; Charleston, M.A.; Duffy, S.; Shapiro, B.; Holmes, E.C. Insights into the Evolutionary History of an Emerging Livestock Pathogen: Porcine Circovirus 2. J. Virol. 2009, 83, 12813–12821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.J.; Gu, J.Y.; Xing, G.; Qiu, X.H.; An, S.T.; Wang, Y.X.; Zhang, C.; Liu, C.M.; Gong, W.J.; Tu, C.C.; et al. Genetic diversity of porcine circovirus type 2 in China between 1999–2017. Transbound. Emerg. Dis. 2019, 66, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Ssemadaali, M.A.; Ilha, M.; Ramamoorthy, S. Genetic diversity of porcine circovirus type 2 and implications for detection and control. Res. Vet. Sci. 2015, 103, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Palinski, R.; Piñeyro, P.; Shang, P.; Yuan, F.; Guo, R.; Fang, Y.; Byers, E.; Hause, B.M. A novel porcine circovirus distantly related to known circoviruses is associated with porcine dermatitis and nephropathy syndrome and reproductive failure. J. Virol. 2016, 91, e01879-16. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; He, W.; Zhu, H.; Bi, Y.; Wang, R.; Xing, G.; Zhang, C.; Zhou, J.; Yuen, K.Y.; Gao, G.F.; et al. Origin, Genetic Diversity, and Evolutionary Dynamics of Novel Porcine Circovirus 3. Adv. Sci. 2018, 5, 1800275. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, T.; Niu, G.; Liu, X.; Zhang, X.; Zhang, Y.; Ren, L. Recent progress on porcine circovirus type 3. Infect. Genet. Evol. 2019, 73, 227–233. [Google Scholar] [CrossRef]
- Sun, J.; Wei, L.; Lu, Z.; Mi, S.; Bao, F.; Guo, H.; Tu, C.; Zhu, Y.; Gong, W. Retrospective study of porcine circovirus 3 infection in China. Transbound. Emerg. Dis. 2018, 65, 607–613. [Google Scholar] [CrossRef]
- Ku, X.; Chen, F.; Li, P.; Wang, Y.; Yu, X.; Fan, S.; Qian, P.; Wu, M.; He, Q. Identification and genetic characterization of porcine circovirus type 3 in China. Transbound. Emerg. Dis. 2017, 64, 703–708. [Google Scholar] [CrossRef]
- Li, G.R.; Wang, H.J.; Wang, S.L.; Xing, G.; Zhang, C.; Zhang, W.Y.; Liu, J.; Zhang, J.Y.; Su, S.; Zhou, J.Y. Insights into the genetic and host adaptability of emerging porcine circovirus 3. Virulence 2018, 9, 1301–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzo, G.; Legnardi, M.; Hjulsager, C.K.; Klaumann, F.; Larsen, L.E.; Segales, J.; Drigo, M. Full-genome sequencing of porcine circovirus 3 field strains from Denmark, Italy and Spain demonstrates a high within-Europe genetic heterogeneity. Transbound. Emerg. Dis. 2018, 65, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Berg, M.; Fossum, C.; Wallgren, P.; Blomstrom, A.L. Detection and genetic characterisation of porcine circovirus 3 from pigs in Sweden. Virus Genes 2018, 54, 466–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadejek, T.; Wozniak, A.; Milek, D.; Biernacka, K. First detection of porcine circovirus type 3 on commercial pig farms in Poland. Transbound. Emerg. Dis. 2017, 64, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.J.; McKillen, J.; Allan, G. Porcine circovirus type 3 in the UK. Vet. Rec. 2017, 181, 599. [Google Scholar] [CrossRef]
- Tochetto, C.; Lima, D.A.; Varela, A.P.M.; Loiko, M.R.; Paim, W.P.; Scheffer, C.M.; Herpich, J.I.; Cerva, C.; Schmitd, C.; Cibulski, S.P.; et al. Full-Genome Sequence of Porcine Circovirus type 3 recovered from serum of sows with stillbirths in Brazil. Transbound. Emerg. Dis. 2018, 65, 5–9. [Google Scholar] [CrossRef]
- Zheng, S.; Shi, J.; Wu, X.; Peng, Z.; Xin, C.; Zhang, L.; Liu, Y.; Gao, M.; Xu, S.; Han, H.; et al. Presence of Torque teno sus virus 1 and 2 in porcine circovirus 3-positive pigs. Transbound. Emerg. Dis. 2018, 65, 327–330. [Google Scholar] [CrossRef]
- Franzo, G.; Tucciarone, C.M.; Drigo, M.; Cecchinato, M.; Martini, M.; Mondin, A.; Menandro, M.L. First report of wild boar susceptibility to Porcine circovirus type 3: High prevalence in the Colli Euganei Regional Park (Italy) in the absence of clinical signs. Transbound. Emerg. Dis. 2018, 65, 957–962. [Google Scholar] [CrossRef]
- Klaumann, F.; Dias-Alves, A.; Cabezon, O.; Mentaberre, G.; Castillo-Contreras, R.; Lopez-Bejar, M.; Casas-Diaz, E.; Sibila, M.; Correa-Fiz, F.; Segales, J. Porcine circovirus 3 is highly prevalent in serum and tissues and may persistently infect wild boar (Sus scrofa scrofa). Transbound. Emerg. Dis. 2019, 66, 91–101. [Google Scholar] [CrossRef]
- Sun, W.; Wang, W.; Xin, J.; Cao, L.; Zhuang, X.; Zhang, C.; Zhu, Y.; Zhang, H.; Qin, Y.; Du, Q.; et al. An epidemiological investigation of porcine circovirus 3 infection in dogs in the Guangxi Province from 2015 to 2017, China. Virus Res. 2019, 270, 197663. [Google Scholar] [CrossRef]
- Yue, F.X. Establishment and preliminary application of multiplex PCR detection methods for PCV2, PPV, PRV and PRRSV. Chin. Vet. Sci. 2008, 8, 691–696. [Google Scholar]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Lam-Tung, N.; Schmidt, H.A.; Arndt, V.H.; Bui Quang, M. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar]
- Stamatakis, A. RAxML Version 8: A tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Delport, W.; Poon, A.F.Y.; Frost, S.D.W.; Pond, S.L.K. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Pond, S.L.K.; Scheffler, K. FUBAR: A Fast, Unconstrained Bayesian AppRoximation for Inferring Selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Pond, S.L.K. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Pond, S.L.K. Less Is More: An Adaptive Branch-Site Random Effects Model for Efficient Detection of Episodic Diversifying Selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Ambrish, R.; Alper, K.; Yang, Z. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar]
- Franzo, G.; He, W.T. A Shift in Porcine Circovirus 3 (PCV-3) History Paradigm: Phylodynamic Analyses Reveal an Ancient Origin and Prolonged Undetected Circulation in the Worldwide Swine Population. Adv. Sci. in press.
- He, W.T.; Auclert, L.Z.; Zhai, X.; Wong, G.; Zhang, C.; Zhu, H.; Xing, G.; Wang, S.; He, W.; Li, K.; et al. Interspecies transmission, genetic diversity, and evolutionary dynamics of pseudorabies virus. J. Infect. Dis. 2018, 219, 1705–1715. [Google Scholar] [CrossRef]
- Klaumann, F.; Correa-Fiz, F.; Franzo, G.; Sibila, M.; Nunez, J.I.; Segales, J. Current Knowledge on Porcine circovirus 3 (PCV-3): A Novel Virus with a Yet Unknown Impact on the Swine Industry. Front. Vet. Sci. 2018, 5, 315. [Google Scholar] [CrossRef]
- Zheng, S.; Wu, X.; Zhang, L.; Xin, C.; Liu, Y.; Shi, J.; Peng, Z.; Xu, S.; Fu, F.; Yu, J.; et al. The occurrence of porcine circovirus 3 without clinical infection signs in Shandong Province. Transbound. Emerg. Dis. 2017, 64, 1337–1341. [Google Scholar] [CrossRef]
- Franzo, G.; Legnardi, M.; Tucciarone, C.M.; Drigo, M.; Klaumann, F.; Sohrmann, M.; SegalEs, J. Porcine circovirus type 3: A threat to the pig industry? Vet. Rec. 2018, 182, 83. [Google Scholar] [CrossRef]
- Saraiva, G.L.; Vidigal, P.M.P.; Fietto, J.L.R.; Bressan, G.C.; Silva Junior, A.; de Almeida, M.R. Evolutionary analysis of Porcine circovirus 3 (PCV3) indicates an ancient origin for its current strains and a worldwide dispersion. Virus Genes 2018, 54, 376–384. [Google Scholar] [CrossRef]
- Franzo, G.; Cortey, M.; Segales, J.; Hughes, J.; Drigo, M. Phylodynamic analysis of porcine circovirus type 2 reveals global waves of emerging genotypes and the circulation of recombinant forms. Mol. Phylogenet. Evol. 2016, 100, 269–280. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Zhao, J.; Xing, G.; Li, G.; Wang, R.; Wang, Z.; Zhang, C.; Franzo, G.; Su, S.; Zhou, J. Genetic analysis and evolutionary changes of Porcine circovirus 2. Mol. Phylogenet. Evol. 2019, 139, 106520. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Amplicon Length (bp) | |
|---|---|---|
| PCV3-D-F | ACTTAGAGAACGGACTTGTAACGAA | 649 |
| PCV3-D-R | AAATGAGACACAGAGCTATATTCAG | |
| PCV3-1-F | ATTATGGATGCTCCTCATCGTG | 553 |
| PCV3-1-R | CATCTTCTCCGCAACTTCAGTC | |
| PCV3-2-F | GACTGAAGTTGCGGAGAAGATG | 789 |
| PCV3-2-R | CGGCACGAAAGAAGTTTGGATT | |
| PCV3-3-F | CCCACATGCGAGGGCGTTTACC | 895 |
| PCV3-4-R | CGAGGCCGCTTCATCATCCACT | |
| PCV2-D-F | AGAAGCTCTCTATCGGAG | 569 |
| PCV2-D-R | AAGGTTGAATTCTGGCCC | |
| CSFV-D-F | TAGGGTGGACGGGTGTCATAGAGT | 566 |
| CSFV-D-R | AAGCATATATTGCTGGAAGTAGCT | |
| PRRSV-D-F | GCCTCGTGTTGGGTGGCAGAA | 532 |
| PRRSV-D-R | CGCCCTAATTGAATAGGTGACTT |
| Isolated Strains | Sequence Similarity among Isolated Strains | Sequence Similarity between Isolated Strains and Reference Strain |
|---|---|---|
| Rep gene (nt) | 99.0–100% | 98.2–100% |
| Rep gene (aa) | 99.3–100% | 97.2–100% |
| Cap gene (nt) | 98.6–99.8% | 97.8–100% |
| Cap gene (aa) | 98.1–100% | 96.7–100% |
| Full-length (nt) | 98.85–99.80% | 97.26–99.90% |
| AA | FEL | SLAC | FUBAR | MEME | ||||
|---|---|---|---|---|---|---|---|---|
| dN-dS | p-Value | dN-dS | p-Value | dN-dS | Post.Pro | w+ | p-Value | |
| 122 | 6.515 | 0.002 | 13.8 | 0.00217 | 11.731 | 0.999 | >100 | 0 |
| 320(24) | 2.784 | 0.027 | 6.29 | 0.0585 | 4.93 | 0.993 | >100 | 0.02 |
| 373(77) | 3.44 | 0.039 | 5.02 | 0.285 | 4.164 | 0.982 | >100 | 0.1 |
| 352(56) | 3.739 | 0.055 | 5.66 | 0.265 | 5.578 | 0.987 | >100 | 0.12 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Xu, Q.; Chen, H.; Luo, X.; Wu, Q.; Tan, C.; Pan, Q.; Chen, J.-L. Evolution and Genetic Diversity of Porcine Circovirus 3 in China. Viruses 2019, 11, 786. https://doi.org/10.3390/v11090786
Chen Y, Xu Q, Chen H, Luo X, Wu Q, Tan C, Pan Q, Chen J-L. Evolution and Genetic Diversity of Porcine Circovirus 3 in China. Viruses. 2019; 11(9):786. https://doi.org/10.3390/v11090786
Chicago/Turabian StyleChen, Ye, Quanming Xu, Hong Chen, Xian Luo, Qi Wu, Chen Tan, Qidong Pan, and Ji-Long Chen. 2019. "Evolution and Genetic Diversity of Porcine Circovirus 3 in China" Viruses 11, no. 9: 786. https://doi.org/10.3390/v11090786
APA StyleChen, Y., Xu, Q., Chen, H., Luo, X., Wu, Q., Tan, C., Pan, Q., & Chen, J. -L. (2019). Evolution and Genetic Diversity of Porcine Circovirus 3 in China. Viruses, 11(9), 786. https://doi.org/10.3390/v11090786