1. Introduction
Coronaviruses (CoVs) are classified into four genera based on phylogeny: alpha-CoV, beta-CoV, gamma-CoV, and delta-CoV. Avian infectious bronchitis virus (IBV) is a major species coronavirus in the gamma-CoV group, causing infectious bronchitis (IB) and impairing the respiratory, reproductive, digestive, and even nervous systems of poultry of all ages and different species [
1]. Due to viral variations, vaccinations are not always effective. At present, IBV has spread worldwide, and IB is one of the most difficult-to-control infectious diseases in the poultry industry [
2].
As one of the members of the CoVs, IBV possesses a single-stranded, positive-sense RNA genome, which comprises two overlapping open reading frames (ORFs) encoding polyproteins 1a and 1ab, the main structural proteins spike (S), envelope (E), membrane (M), and nucleocapsid (N). The viral S protein is the major surface protein of IBV, which is cleaved into S1 and S2 subunits by the host serine protease furin [
3]. The S1 protein is associated with viral antigenicity and tissue tropism of the virus and plays vital roles in the induction of neutralizing antibodies and attachment to host cell receptors [
3]. The interaction between viral and host proteins is an important means for viruses to successfully infect and replicate within host cells. However, few IBV strains can infect avian passage cell lines, which seriously hinders the progress of basic research on IBV pathogenesis.
Tracheal ring organ culture (TOC) is a commonly used and reliable method for isolating and culturing IBV, as an alternative to embryonated chicken eggs. IBV field isolates that do not replicate in primary cell culture can replicate and proliferate in TOC without previous adaptation in embryonated chicken eggs [
4]. With the exception of a few reports using TOC as an in vitro model to study IBV pathogenicity [
5,
6], the application of TOC is limited in the isolation of field isolates. Therefore, it is of great significance to establish a TOC system to conduct host protein identification and IBV-host interactions.
The interaction between viral and host proteins is one of the most important routes for viruses to successfully infect host cells and carry out replication. Heat shock proteins (HSPs), including heat shock protein 70 (HSP70), are a large family of chaperone proteins found in most eukaryotes and bacteria. HSPs are responsible for the correct folding of proteins, protecting cells from stress, stimulating the production of immune and inflammatory factors, and regulating the host immune response. During the course of viral infection, it is utilized by the virus to complete the viral life cycle at all stages and to adapt to and escape the host’s antiviral response [
7].
HSP70 is a receptor or coreceptor of various viruses, including dengue virus, Japanese encephalitis virus, and rotavirus [
8,
9,
10]. To date, the functional cellular receptor for IBV infection is unknown. In addition, whether host HSP70 is associated with IBV infection awaits elucidation. In this study, HSP70 was found to be one of the predicted IBV S1-interacting candidates by LC-MS/MS analysis and was subjected to further confirmation.
2. Materials and Methods
2.1. Virus, Cells, Antibodies, Plasmids, and SPF Embryonated Chicken Eggs
IBV reference strains Beaudette (GI-1) and M41 (GI-1) as well as field isolates GX191219 (GI-7), QD191227 (GI-13), HF200702 (GI-19), and YC181031 (GI-22) used in this study were stored in our laboratory and were genotyped according to the typing method established by Valastro et al. [
11]. Vero and 293T cells were maintained in DMEM supplemented with 10% FBS at 37 °C in a 5% CO
2 atmosphere. Chicken embryonic kidney (CEK) cells were prepared as previously described [
8]. In-house mAbs against IBV S1 (1H1), M (2B3) and N (4H4) proteins, and rabbit polyclonal antibodies against HSP70 were produced by our research group. Eukaryotic expression plasmids pCMV-Flag-N-HSP70 and pCMV-Myc-N-BD-S1were constructed by our group. Specific pathogen-free (SPF) embryonated chicken eggs were purchased from Ningbo Chunpai Agricultural Technology Company (Ningbo, China).
2.2. Preparation of Tracheal Ring Organ Culture
Tracheas were removed from 18 to 20 days old embryonated chicken eggs and placed into petri dishes containing DMEM. Fats and surrounding connective tissues were discarded as much as possible, and the lumen was rinsed 5–10 times using a syringe filled with DMEM. The cleaned tracheas were transversely sectioned into 0.5–1 mm tracheal rings with small sharp scissors. The rings were then washed twice with culture medium and placed into 24-well plates with 4 rings in each well containing 0.5 mL TOC medium (DMEM supplemented with 5% FBS and 0.1% antibiotic (containing 100 i.u./mL penicillin and 100 ig/mL streptomycin)) and cultured in a 37 ℃ cell incubator with 5% CO2. After culturing for 12–24 h, tracheal rings with active ciliary beating were used for infection.
2.3. Determination of TOC-ID50 of IBV Viruses
The TOC-ID50 of the IBV virus strain was measured using tracheal rings with vigorous beating cilia after a 24 h preparation. Serial dilutions of IBV-infected chicken embryo allantoic fluid ranging from 10−1 to 10−10 were inoculated into the tracheal rings in the wells of the 24-well plates, with 5 wells for each dilution, followed by incubation at 37 °C with 5% CO2 for 1 h. Next, after 3 washes with PBS, the tracheal rings were cultured with TOC medium in the incubator for 144 h. During this period, the culture medium was renewed every 48 h. The samples were scored as infection-positive when 70% ciliostasis was observed. The calculation of TOC-ID50 was conducted according to the Reed–Muench method as follows: distance ratio = (positive rate above 50% − 50%)/(positive rate above 50% − positive rate below 50%) and lgTOC-ID50 = logarithm of the highest antibody dilution above 50% positive rate + distance ratio × logarithm of the dilution factor.
2.4. Infection of TOC with IBV
Freshly prepared TOCs with vigorously beating cilia were infected with different IBV strains at a dose of 1000 TOC-ID50, and uninfected TOCs were used as controls. Successful infection was confirmed by RT-PCR using one set of primers targeting the IBV N gene (IBV-N-F: 5′-GATGGTAATTTCCGTTGGGA-3′/IBV-N-R: 5′-CATTGTTCCTCTCCTCATCT-3′). Briefly, 3 tracheal rings in 500 μL PBS were processed using a tissue crusher for 1 min, followed by 3 rounds of repeated freezing and thawing, and the supernatant was subjected to total RNA extraction using TRIzol reagent. The RNA was then transcribed into cDNA according to the instructions provided by the Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, MA, USA). PCR was performed in a 25 μL reaction containing 12.5 μL of 2× PCR mixture, 0.5 μL of each primer (10 μM), and 1.5 μL of cDNA (10 ng/μL), then subjected to the amplification programs at 95 °C for 5 min, 35 cycles of denaturation at 94 °C for 30 s, annealing at 50 °C for 30 s, an extension at 72 °C for 30 s, and a final extension of 72 °C for 10 min. Real-time qRCR (RT-qPCR) was performed to detect the relative abundance of IBV-N transcripts using the ChamQ Universal SYBR qPCR master mix (Vazyme Biotechnology, Nanjing, China) in a LightCycler 96 sequence detector system (Roche Diagnostics, Mannheim, Germany). A grading system of 0 (100% ciliostasis) to 4 (0% ciliostasis) was used to score the ciliary activity of the TOC. The activity of control tracheal rings at 12 hpi was recorded at the highest level of “4”, whereas the scores in the other groups ranged from 0 to 4 at the different infection time points, respectively. For further analysis, the dynamics of IBV infection in TOC were monitored via 6 samples for each time point with 3 rings for HE, IHC, and IFA assays, with the remaining 3 were used for Western blot analysis.
2.5. HE, IHC and IFA
Three tracheal rings in each tube were washed once with 4% paraformaldehyde (PFA), then kept in 1 mL 4% PFA and sent to Nanjing Freethinking Biotechnology Co., Ltd. (Nanjing, China) to perform hematoxylin-eosin (HE) staining and immunohistochemistry (IHC) assay.
2.6. Immunoprecipitation and Co-Immunoprecipitation Assays
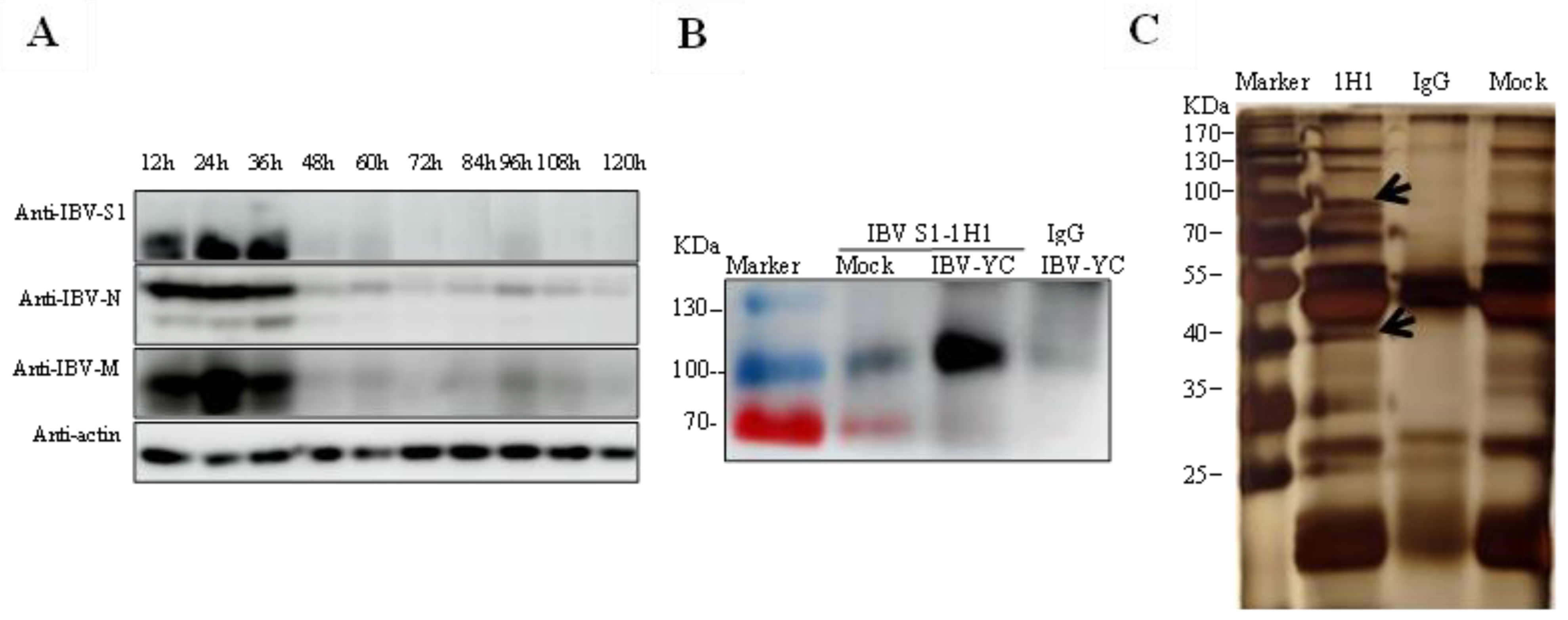
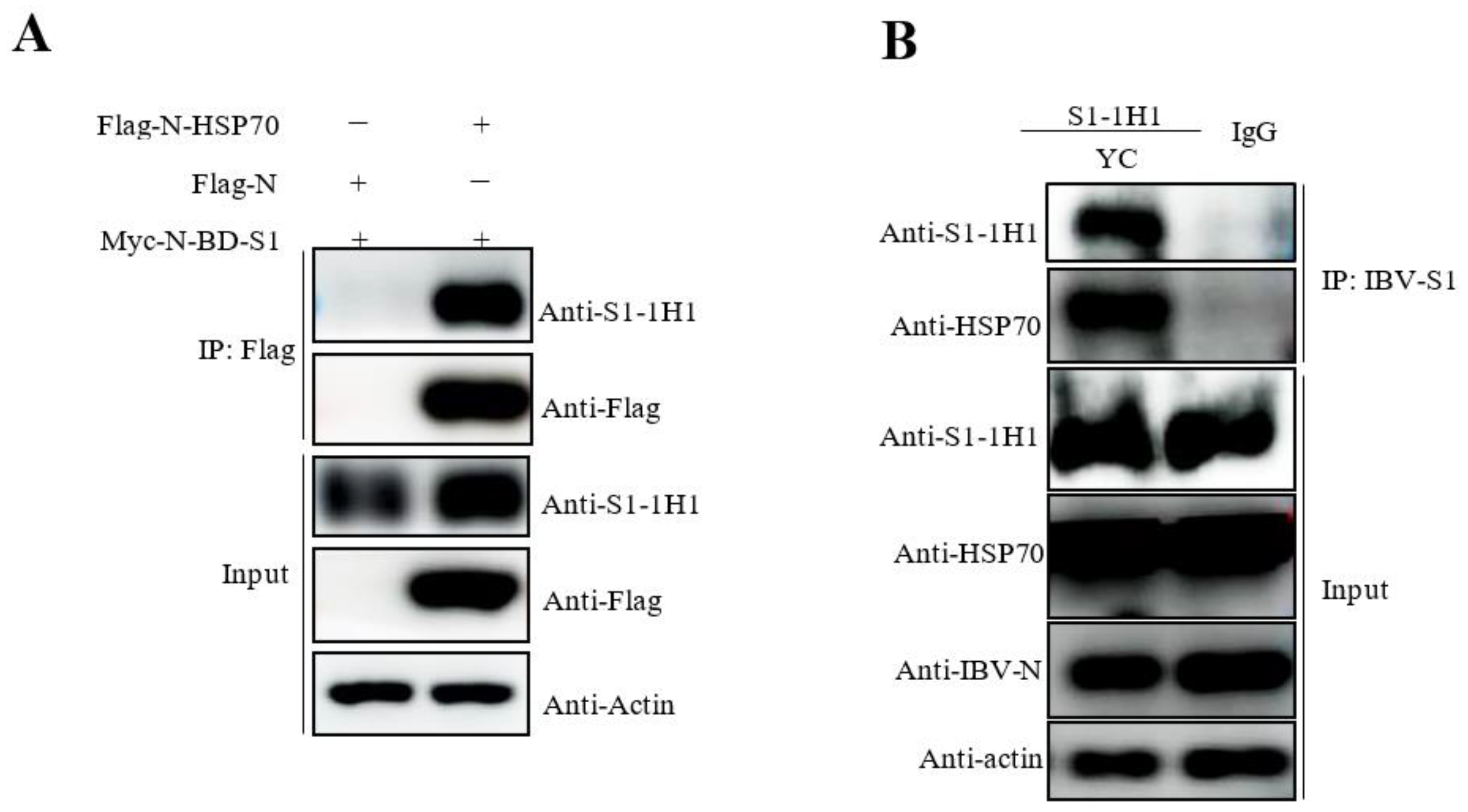
The membrane proteins from IBV YC181031 infected TOCs were extracted using the Membrane Protein Extraction Kit (Beyotime, Shanghai, China) following the manufacturer’s protocol. Membrane protein extracts were immunoprecipitated (IP) with mAb 1H1 against IBV S1 protein and a mAb against mouse IgG (A7028, Abcam, Cambridge, UK). The precipitated proteins were detected by silver staining and Western blot analyses using anti-S1 and anti-HSP70 antibodies. Next, 293T cells were cotransfected with pCMV-Flag-N-HSP70/Myc-N-BD-S1, and the Flag-tagged HSP70 was immunoprecipitated with Flag antibodies and captured by Sepharose A/G beads. The precipitated proteins were then detected by Western blot analysis using anti-Flag antibodies and mAb 1H1 to IBV S1.
2.7. SDS-PAGE, Western Blot Analysis and Silver Staining
TOC lysates and immunoprecipitated proteins were separated using SDS-PAGE and transferred onto a nitrocellulose membrane. After blocking with 5% skim milk, the membranes were incubated with the indicated primary antibodies at 4 °C overnight. After three washes with PBS, the blots were incubated with HRP-labeled anti-mouse/rabbit IgG at 4 °C for 1 h. Protein bands were then visualized using an enhanced chemiluminescence reagent and imaged using an AI680 Imager (GE Healthcare, Chicago, IL, USA).
The polyacrylamide gel was fixed with a 4:1:5 ratio of ethanol, glacial acetic acid, and water at room temperature for 2 h and then washed thrice with water. After incubation with a sensitizing solution containing sodium acetate, sodium thiosulfate, and absolute ethanol, the gel was transferred to the silver stain solution and incubated in the dark at room temperature for 30 min. Finally, a color developer containing anhydrous sodium carbonate, formaldehyde, and sodium thiosulfate was used and incubated in the dark with the gel. The reaction was terminated by adding 5% glacial acetic acid. The stained bands were imaged using an ImageScanner III instrument, and the bands of interest were excised from the gel for liquid chromatography-mass spectrometry (LC-MS) analysis.
2.8. Mass Spectrometry and Analysis
Three independent immunoprecipitation experiments to identify the host proteins interacting with IBV S1 were performed. After silver staining detection, specific bands in the IBV S1 mAb group as compared to the control IgG group were collected from three independent gels and sent for commercial mass spectrometry (LC-MS/MS) analysis carried out by Shanghai Applied Protein Technology Company. Protein searches were carried out using Mascot 2.2 software. Protein identification was performed using the following criteria: (a) trypsin-digested peptides with 2 max missed cleavages allowed, (b) proteomics tools: 3.1.6, (c) >1 unique peptides, (d) filter by score ≥20. Proteins found in the respective negative control samples were eliminated from the dataset to remove nonspecific interactions. Proteins represented by at least two unique peptides were considered for further analysis, and the proteins in the negative control were excluded.
Candidate proteins from LC–MS/MS analysis were subjected to GO annotation and KEGG analysis by using the DAVID website (
https://david.ncifcrf.gov/home.jsp, accessed on 17 March 2021) under the three major categories of biological processes (BO), molecular functions (MF), and cell components (CC).
2.9. Infection of Vero Cells Overexpressing HSP70 with the IBV Beaudette Strain
The plasmid pCMV-Flag-N-HSP70 was transfected into Vero cells using the jetPRIME kit (Polyplus). The cells were infected with the IBV Beaudette strain (MOI = 1) 24 h after transfection and incubated at 37 °C for 12 h, 24 h, and 36 h, respectively. The cells were harvested after three washes with PBS and subjected to Western blot analysis.
4. Discussion
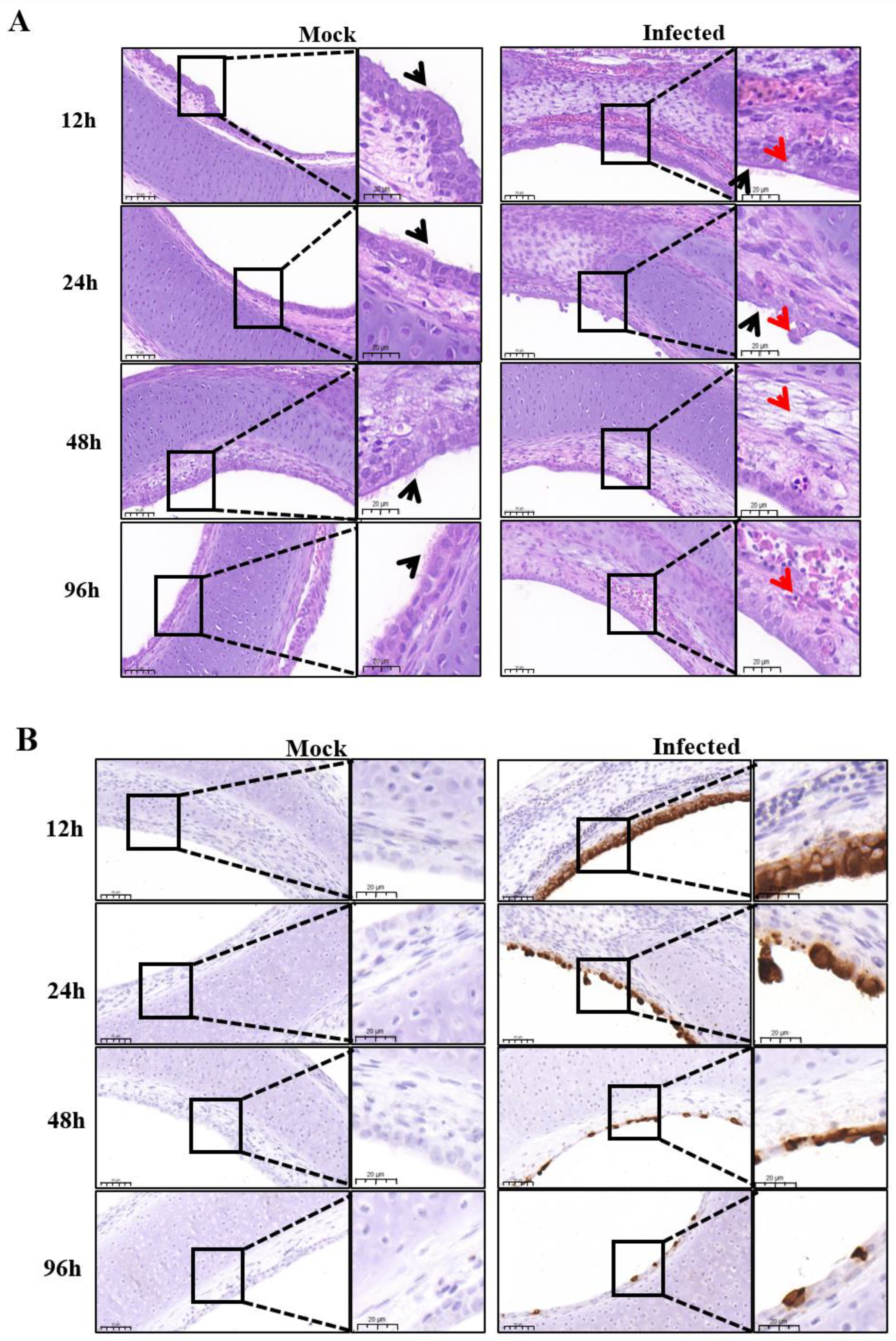
In this study, one IBV reference strain M41 and four isolate strains (GX191219, QD191227, HF200702, and YC181031) were used to infect the tracheal rings of chicken embryos, respectively. The degree of ciliostasis was used to evaluate the activity of tracheal tissues infected with IBV [
4]. All selected strains could cause ciliostasis without any previous adaptation. Cilial activity of the tracheas in all infected groups maintained for 2 days after infection, then decreased at 2–4 hpi accompanied by gradual loss of cilia, followed by complete ciliostasis at 144 hpi (
Figure 1A). The ciliary activity of YC101031 infected tracheas seemed to decrease more at 96 hpi (
Figure 1A) than those in the other infected groups, suggesting different susceptibilities among strains towards tracheal damage. We did not perform a pathogenicity comparison study among the selected strains. In a previous study, we found that the vaccine strain H120 causes complete ciliostasis 72 hpi earlier than that of some field isolates. Therefore, it is difficult to comment on whether the degree of damage to tracheal rings by the virus is related to its virulence. This requires more experimental data.
The selected virus replicated in epithelial cells very well. As shown by IHC analysis, almost every epithelial cell was infected by the virus at 12 hpi (
Figure 2B), while ciliary beating was still very active at 12 hpi, as observed with microscopy (
Figure 1A). In this case, the detection of IBV infection by IHC was more sensitive than ciliostasis observation. Microscopic observation revealed that the cilia of the tracheal ring remained active 24 h after infection (
Figure 1A). However, at this time point, some epithelial cells dropped off the rings, leading to a decrease of virus staining cells in the infected trachea at 24 hpi as compared to that at 12 hpi, as detected by IHC (
Figure 2B). As infection progressed, more ciliated epithelial cells of the tracheas became necrotic and shed from the tracheas at 48 hpi, and by 96 hpi, only a few infected epithelial cells remained, with cilia completely disappearing (
Figure 2A,B).
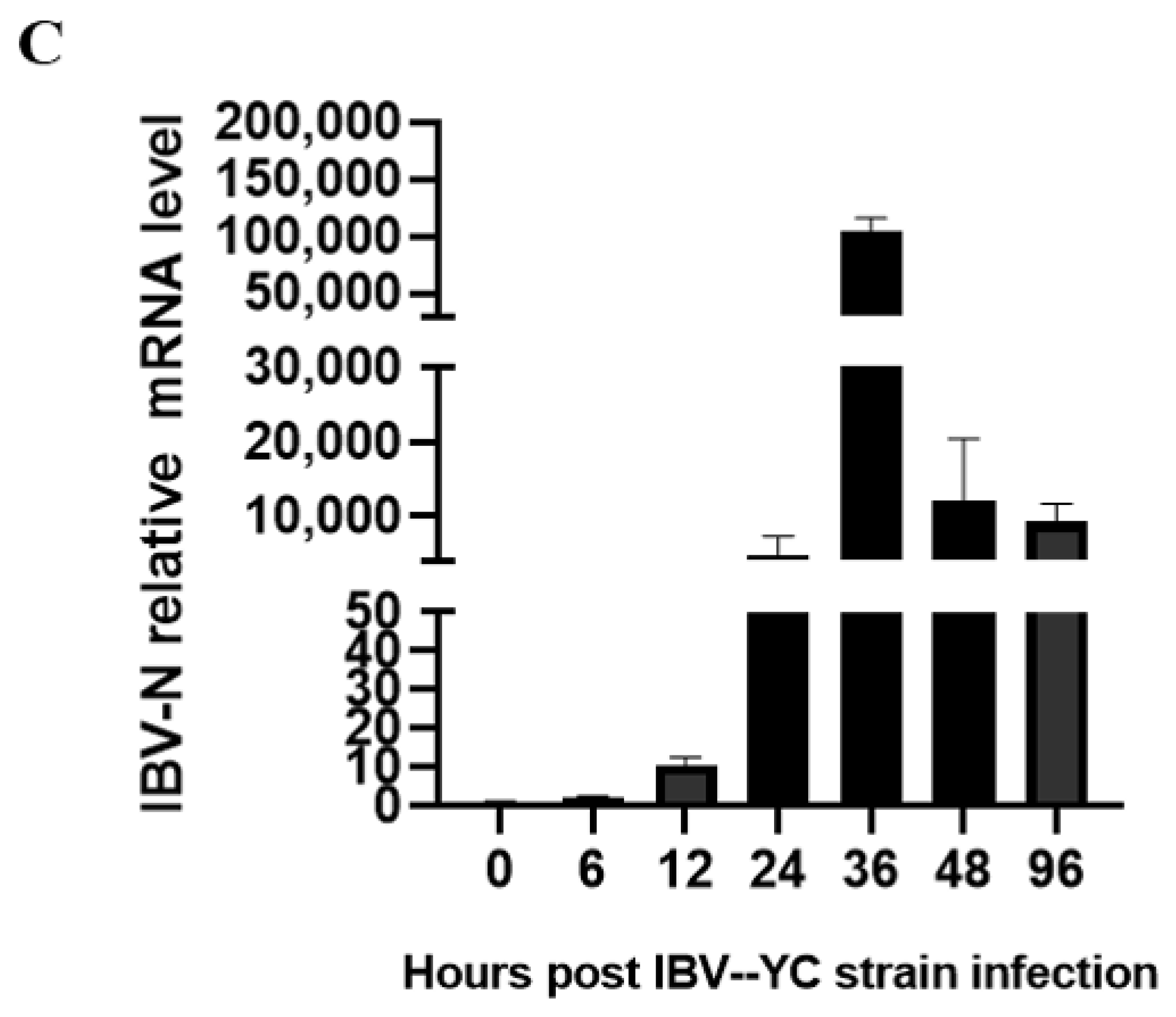
Using IHC analysis, we clearly noticed epithelial cells stained by IBV mAb at 12 hpi, after which the number of stained cells decreased (
Figure 2B). However, by RT-qPCR (
Figure 2C) and Western blot (
Figure 3A) analysis, the highest mRNA expression level and highest protein expression level were observed at 36 hpi and at 24–36 hpi, respectively (
Figure 2C and
Figure 3A). We think this discrepancy is due to the infected cells dropping off the tracheas gradually, but the epithelial cells that had dropped off were collected and subjected to the Western blot and RT-qPCR analysis. Therefore, the TOCs infected at 36 hpi were selected for the study of virus-host interactions by immunoprecipitation.
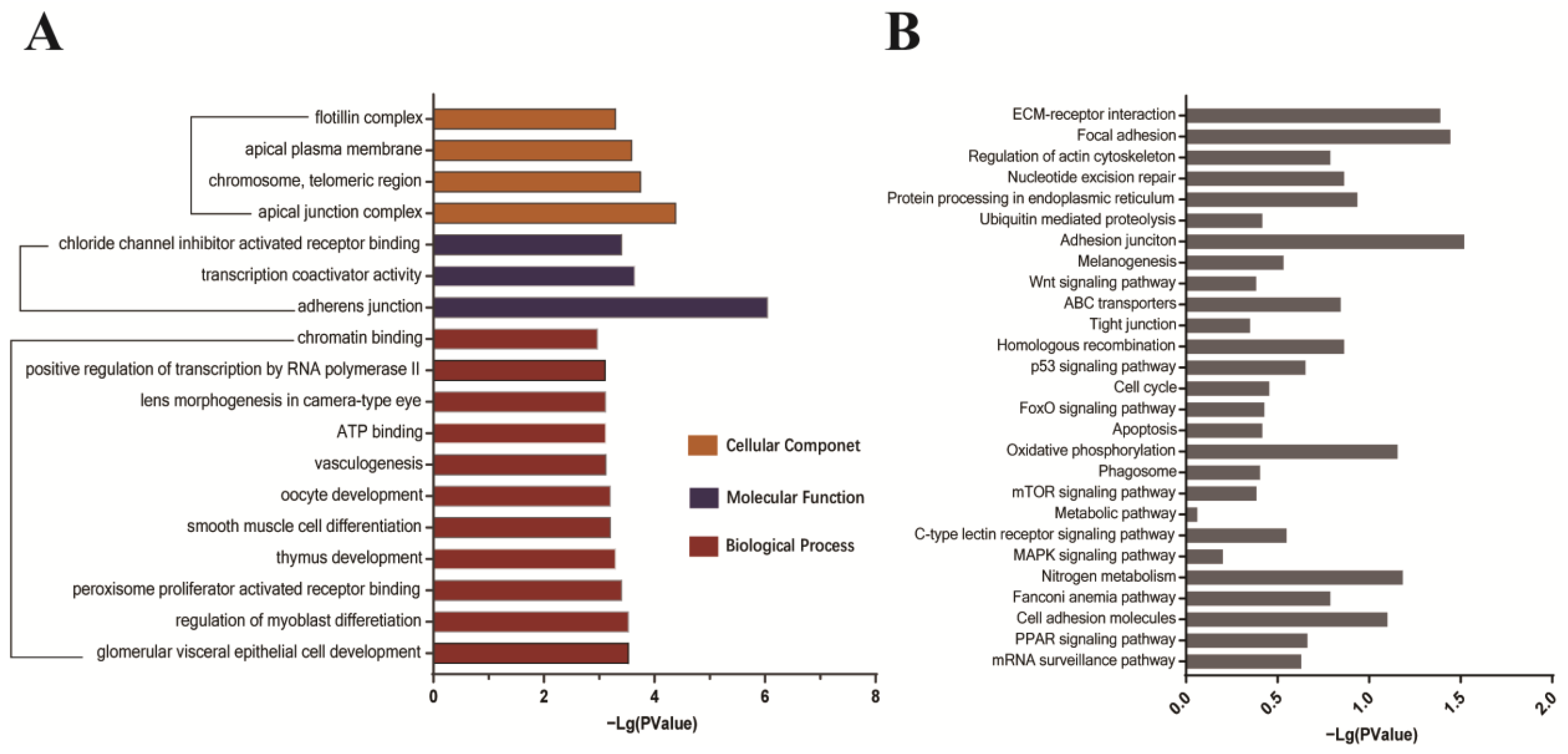
GO and KEGG analyses were performed on the identified candidate proteins. Enrichment analysis results showed that the host proteins interacting with the S1 protein are closely related to cell adhesion and connection, protein metabolism, and transport processes and participate in receptor interaction, apoptosis, and autophagy signaling pathways (
Figure 4A,B and
Supplementary Tables S2 and S3). Among the identified candidates, several proteins such as integrin, beta-catenin, nectin, otogelin, and shroom3 belong to adhesive components [
12,
13,
14], which are potentially important factors for host cell attachment and infection progression during IBV infection.
A previous study identified heat shock protein 70 (HSP70) from the lungs and kidneys of IBV-infected chickens as a binding-associated protein to IBV S1 of the SCZJ3 strain, and infection of CEK cells by SCZJ3 was found to be inhibited by anti-HSP70 antibodies, indicating that HSP70 is a part of the IBV receptor complex [
15]. HSP70 translocates to the membrane of host cells under certain conditions such as heat stress [
16], tumor [
17], or viral infection [
18]. There is evidence that HSP70 serves as a receptor for several viruses [
18,
19,
20]. In our current mass spectrometry data, we also identified HSP70 as a predicted binding-associated protein to S1 of YC181031. Importantly, immunoprecipitation and Western blot analyses showed that the IBV S1 protein of YC181031 is able to interact with HSP70 in TOCs and 293T cells (
Figure 5).
We further investigated the effects of HSP70 on IBV replication by using knockdown and overexpression assays. However, we could not detect clear changes in HSP70 expression by transient transfection of HSP70 interference or overexpression plasmids using the TOC system (data not shown). Additional factors present in TOC may influence the knockdown and overexpression of host genes. In addition, due to the high inherent expression level of native HSP70 in the trachea organ, it may not be easy to interfere with its expression or to observe the effects of HSP70 overexpression upon virus infection. Further fine-tuning or modification of reaction conditions is required in order to use the TOC model to study the functions of host proteins on IBV infection.
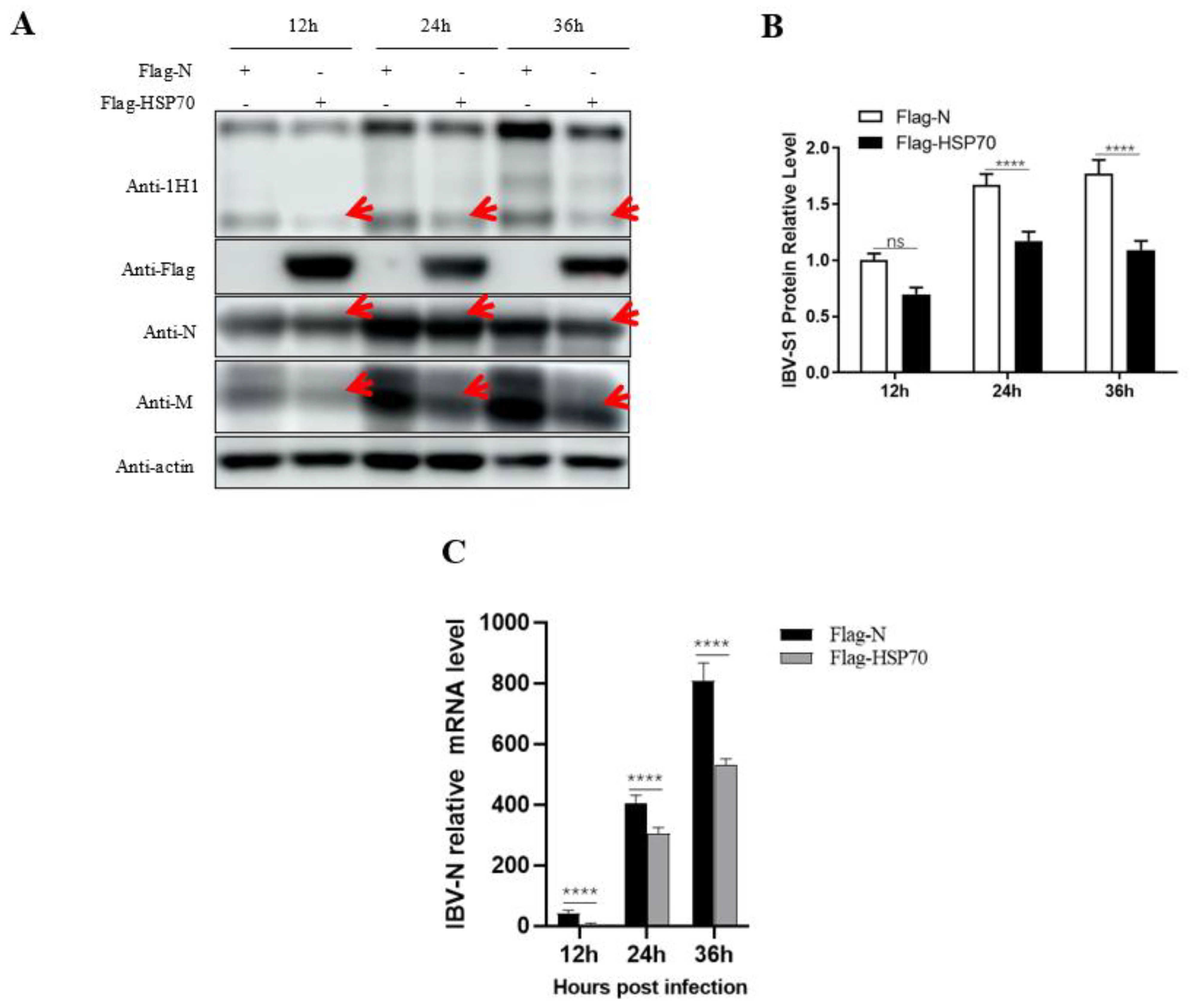
Instead of using TOC, in Vero cells, we observed that HSP70 overexpression inhibited IBV Beaudette growth, indicating that HSP70 might be a potential host anti-viral factor for IBV infection (
Figure 6). This result seems contradictory to a previous report where HSP70 was part of the receptor complex of IBV [
15]. However, HSPs are multifunctional proteins, although the biological functions of HSPs include the maintenance of cellular homeostasis. In cases of viral infection, they can also be employed by viruses to correctly fold their proteins, assist the viruses in completing their life cycles at various stages, help the viruses adapt to and escape from antiviral responses of the host cells, or can be used by the host to combat viral infection, etc. [
7]. We speculate that in the early stage of IBV infection, host HSP70 might assist virus attachment and entry into host cells while inhibiting virus replication after its invasion. The molecular mechanisms of HSP70 in IBV infection will continue to be investigated by our group in the near future.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






