Effect of Host Matrix Molecular Weight on Chain Conformation.
MEH-PPV single molecule emitting forms are first categorized then PL line shapes are next analyzed to determine structural characteristics (i.e., vibrational displacements) of these chromophores.
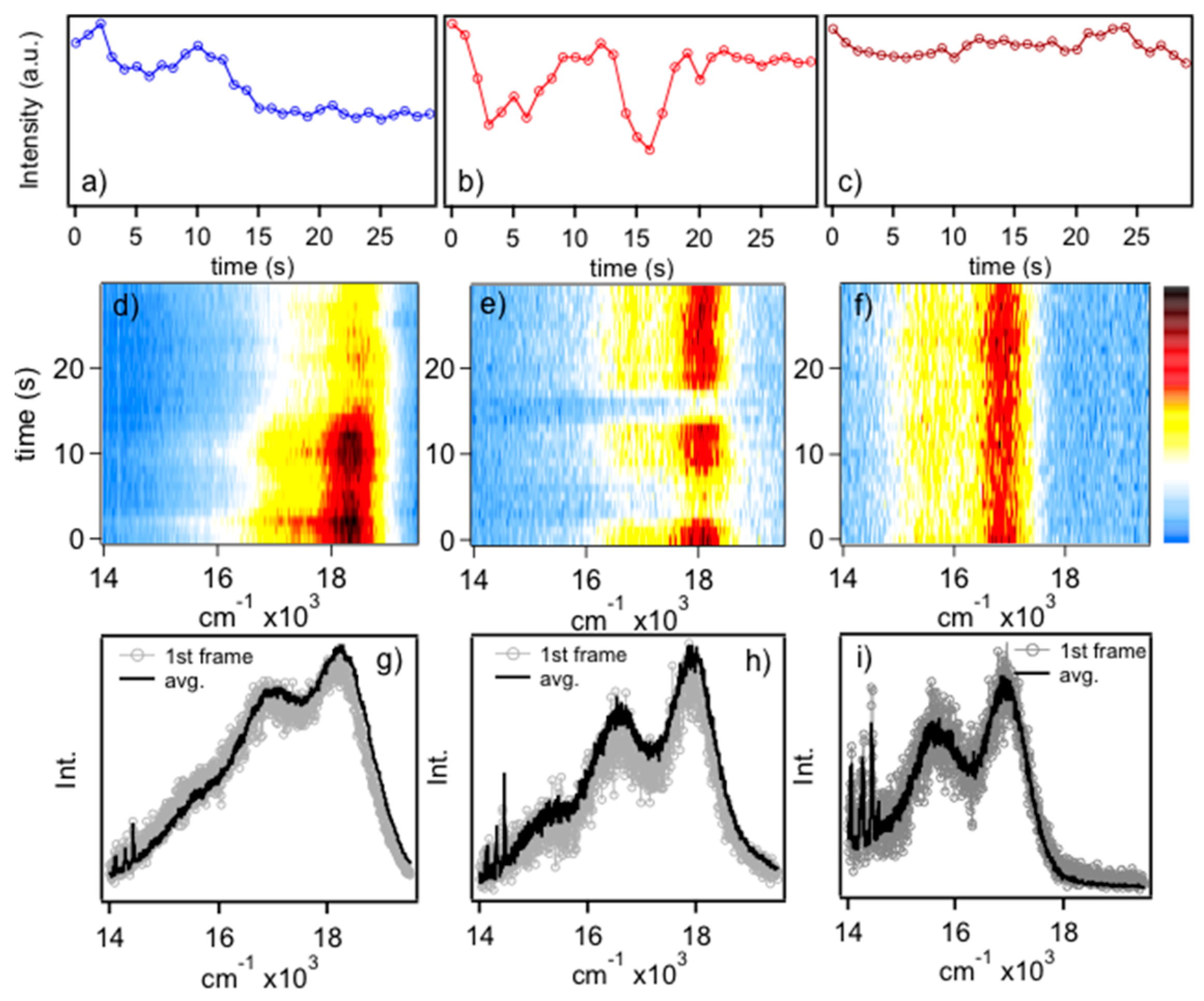
Figure 1 shows representative PL transients (
Figure 1a–c), spectral dynamics (diffusion) (
Figure 1d–f) and comparisons of averaged and first frame spectra (
Figure 1g–i) of MEH-PPV chains dispersed in polystyrene hosts. Spectra can be classified according to their energies, which correspond to blue, red and “ultra-red” emitters and the full distributions are presented and discussed in the following. Assignment of emissive conformers was based on the PL energy of the electronic origin (
E0-0) and sub-ensemble PL spectra were generated by averaging spectra within their respective distributions (vide infra). In general, blue emitters typically show step-wise flickering whereas red emitters show discrete on/off flickering over the timescale of the measurements. These behaviors are indicative of multi-chromophoric and single chromophore emission, respectively, and reflect the conformational characteristics of the individual polymer chains [
11]. Namely, blue emitters originate from solution-like conformations (i.e., defect cylinder) where energy transfer between segments is relatively inefficient probably due to poor dipole–dipole orientation factors. Conversely, red emitting chains exist in collapsed conformations that favor rapid energy migration to as little as a single emitting site and reversible photo-oxidation results in the observed telegraphic (on–off) flickering behavior. As mentioned earlier, these chromophores possess J-aggregate exciton characteristics, which require elongated and planarized segments [
28,
29]. Ultra-red emitters show high photo-stability with no distinct flickering behavior. Very little spectral diffusion is observed for the single molecule spectra in this study, which is expected for the smaller excitation power densities used. It is possible, however, that spectral fluctuations could occur on time scales faster than the PL spectra acquisition times used. However, no significant deviations of energy maxima are observed over the course of the measurement consistent with minor contributions from spectral diffusion. We now consider the structural and conformational characteristics of these emitters and the how they evolve between each form.
There has been ongoing debate over the structural characteristics of the red emitting form of MEH-PPV, which mostly correspond to emissive traps in bulk films (see
Supplementary Materials, Figure S2). Although single molecule spectroscopy studies have shown that this species does not require more than one chain to exist, much of the detailed information in PL line shapes remains to be harvested to construct more meaningful pictures of their structural qualities beyond inferences and expectations from exciton coupling theory. In particular, assessing the extent of geometrical displacements between ground and excited electronic states as well as the vibrations involved is especially important for understanding and controlling charge and energy transfer processes. To this end, we evaluate MEH-PPV single molecules dispersed in polystyrene hosts of varying molecular weight and apply post processing SVA treatments to selectively tune chain conformations. Approximately 50–100 spectra and transients are recorded for each sample and compared by plotting histograms of the energy of the electronic origin transition (
E0-0). This parameter is more meaningful than the peak energy maximum (
Emax) to compare chain conformational characteristics since mixed emitters (i.e., blue and red emitters within the same molecule) tend to bias the distributions typically towards the red emitters. For this reason, we exclude mixed emitters from the analysis, which only comprise about 15% of the total molecules studied. Spectra from each host were recorded under the same conditions and we found no correlation between brightness (integrated PL counts) and type of emitter. This aspect is essential for comparing single molecule statistics since aggregates can often overwhelm single chain emission.
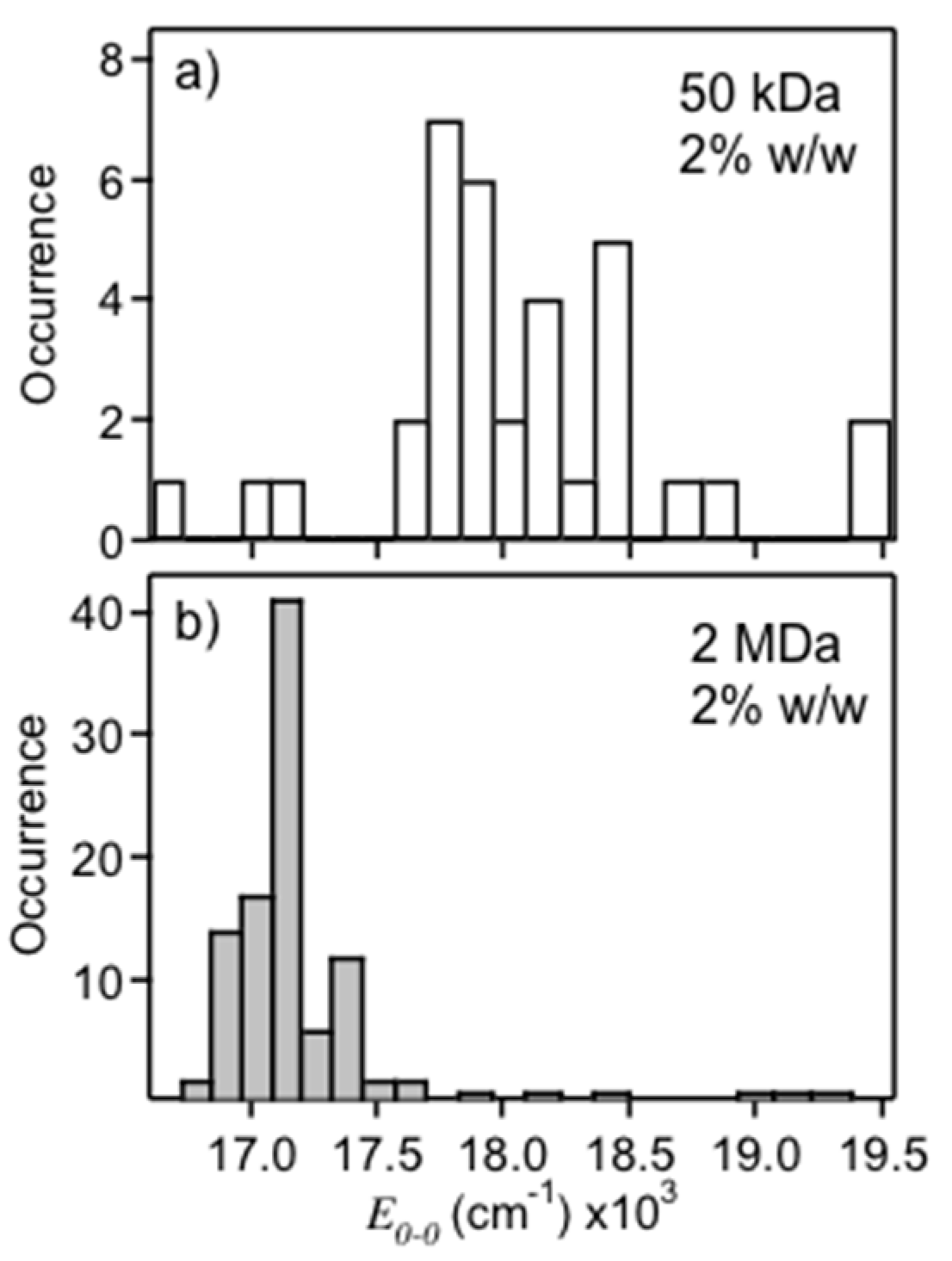
Figure 2 shows PL
E0-0 histograms for MEH-PPV single molecules dispersed in 50 kDa (
Figure 2a) and 2 MDa (
Figure 2b) polystyrene hosts each at a concentration of 2% (
w/
w) in chloroform.
E0-0 distributions of the 50 kDa samples show primarily emission from the blue form (~18,200 cm
−1), whereas the 2 MDa samples show emission exclusively from the red form (~17,100 cm
−1). The spread in PL energies is also narrower for the latter (~100 cm
−1 vs. ~200 cm
−1) indicating less disorder. The ability to select either emitting form likely originates from host–molecule interactions and solvent evaporation times. The number averaged MW of MEH-PPV chains used in this study was ~950 kDa which appears to be better solvated in the lower MW host thus favoring mostly solution-like conformations (blue emitters). Chang et al. also found that high MW MEH-PPV chains are well solvated in small molecule liquid crystals, consistent with our observations [
30]. It is also important to note the viscosity of the 2 MDa polystyrene sample was considerably higher than the 50 kDa sample leading to longer solvent evaporation rates. MEH-PPV chains likely have a longer time to adopt their thermodynamically favorable structure (i.e., collapsed conformations) in these hosts thus mimicking SVA treatments (see below) due to swelling of the matrix.
To test this hypothesis, both polystyrene hosts were cast at lower concentrations (0.2%
w/
w) to produce thinner films (~40 nm).
Figure 3a,b shows
E0-0 histograms for the thin 50 kDa and 2 MDa dispersions. The thinner 50 kDa samples behaved similarly to the thicker films generated, whereas the 2 MDa samples showed considerably more blue emitters in thinner films giving rise to the familiar bimodal emitter distribution [
3,
9]. To determine if the blue emitters could be converted to the red from after casting, samples were subjected to SVA treatments inside a glovebox then sealed with aluminum under a base pressure of ~10
−7 mbar.
Figure 3c,d displays
E0-0 histograms for SVA treated thin films of 2 MDa and 50 kDa samples, respectively. The bimodal distribution collapses to only the red form in both films, similar to the thicker samples of the 2 MDa matrix, indicating that indeed the higher MW hosts (viscosities) have similar outcomes as SVA treatments. Thin annealed samples of the 50 kDa polystyrene host show an overall red-shift due to an increased number of red species, but an additional species emerges approximately 1100 cm
−1 further to the red. A small amount of this form was observed in as-cast films (<10%) that increases up to ~35% upon SVA treatment. These ultra-red emitters appear almost exclusively in the 50 kDa host and may result from surface perturbations with a greater fraction of molecules in contact with the substrate. Previous work from Barnes and coworkers also report a similar species when MEH-PPV molecules are cast directly on glass suggesting a surface-induced folding of the polymer chain [
31,
32]. The similarity of this emitter with bulk films may further indicate these species are aggregates although average intensities were not significantly brighter. We next analyze PL line shapes of each emitting form to generate deeper insights about their structural and conformational attributes and how these affect exciton coupling and energy transfer.
3.1. Extending the MIME Description to Single MEH-PPV Molecules
Although the bimodal emission phenomenon is well-known for single MEH-PPV molecules, surprisingly little attention has been paid to the line shapes which contain useful information surrounding the structural characteristics of chromophores.
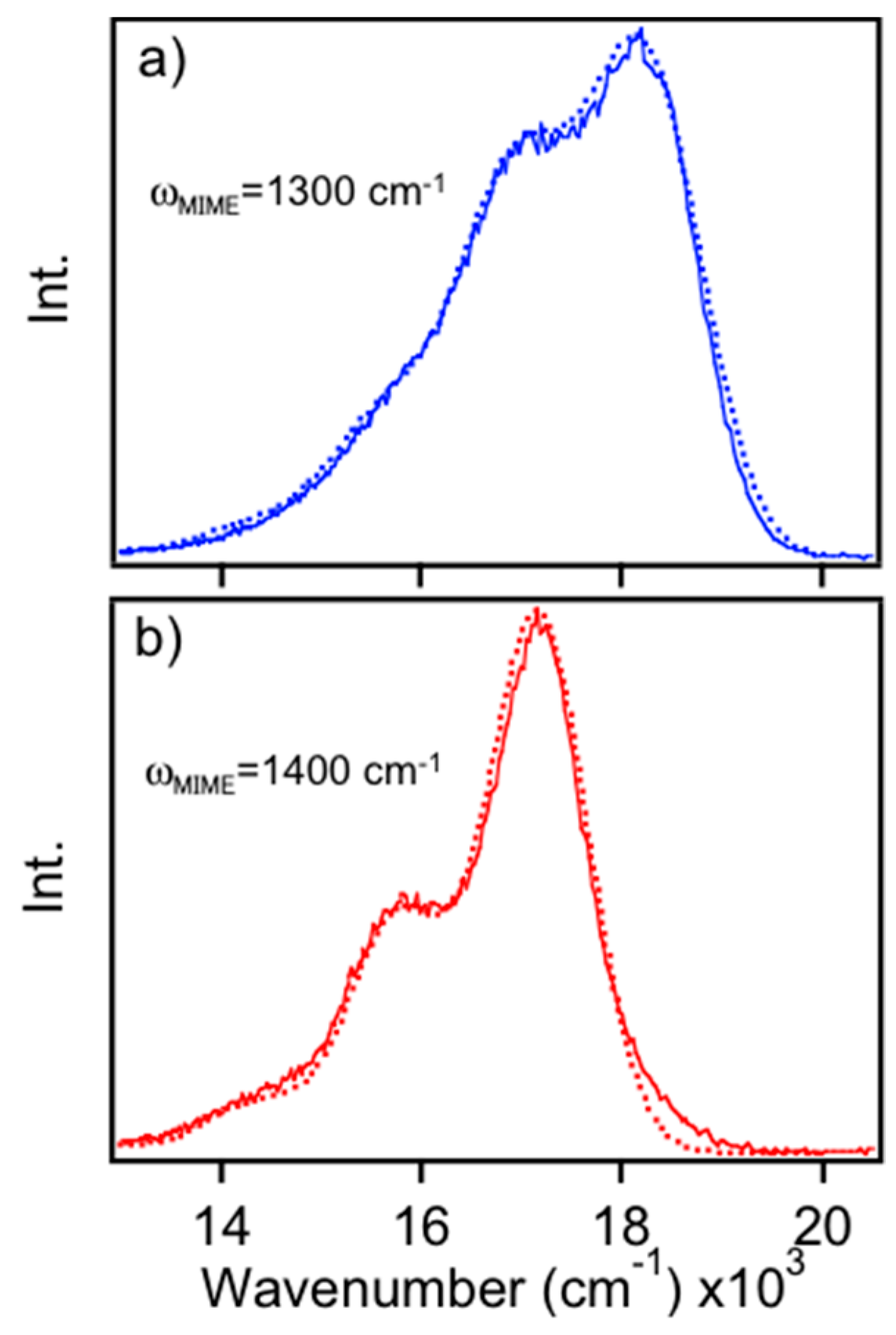
Figure 4a shows sub-ensemble averaged spectra for blue and red emitters generated from thin 2 MDa as-cast films. Similar sub-ensemble averaged spectra are included in the
Supplementary Materials (Figure S3), which were generated from 50 kDa films. To qualitatively illustrate the difference in PL lineshapes for each form, spectra are overlapped at their
E0-0 values. Comparison of these averaged line shapes show noticeable differences, namely, smaller Huang–Rhys factors and vibronic frequency intervals in the red form. These features are consistent with observations of MEH-PPV and related derivatives in solution and solid forms, where the latter always have a larger progression interval. Furthermore, these differences do not arise from significant variations in the refractive index, which is nearly constant in this energy range. The vibronic spectra presented herein display clear manifestations of the MIME where several displaced vibrational modes coalesce into one effective interval. This MIME is more easily observed in individual single molecule spectra since ensemble averaging for each form tends to broaden lineshapes making it less distinct. Analysis of each spectrum can reveal the intrinsic heterogeneity within each emitting form that is otherwise obscured in the ensemble data.
We previously used up to five modes to fit PL spectra of a related derivative, MDMO-PPV, in various forms. It was demonstrated that individual displacements vary substantially depending on the specific sample form, which, in turn, dictate the effective progression interval [
19]. Vibrational displacements were estimated from pre-resonance Raman intensities using the Savin formula,
I1/
I2 = (ω
1Δ
1)
2/(ω
2Δ
2)
2. Importantly, the largest change in displacement between solution and solid forms occurred in the out-of-plane C–H wag mode of the PPV vinylene linkage (~961–966 cm
−1) [
19]. While the relative Raman intensities of this mode are typically small, its displacement, according to the Savin formula, can be comparable or larger than more intense, higher frequency modes (i.e., symmetric CC stretches of the phenyl group, ~1580 cm
−1). To illustrate the effect of varying the displacement of the C–H vinyl wag on the MIME interval, we use a simple two-mode model, namely, the ~1580 and 966 cm
−1 modes, which comprise the upper and lower frequency range from our previous MIME analysis of MDMO-PPV [
19].
PL lineshapes are calculated using the time-dependent theory of spectroscopy of Heller and coworkers [
33]. In this approach, a Gaussian wavepacket (
) is projected vertically from the lowest vibrational level of the initial state (emitting state) to the final state (ground state). The wavepacket evolves in time according to the time-dependent Schrödinger equation. The emission intensity is then described by:
where ω is the frequency of the emitted photon and the multi-mode, time-dependent wavepacket overlap autocorrelation function,
, is given by:
where Δ
k is the dimensionless vibrational displacement for the
k-th mode,
E0-0 is the energy of the electronic origin transition (cm
−1), and Γ is a phenomenological damping factor (cm
−1) that determines observed vibronic linewidths. This expression can be solved analytically provided that the ground and emitting state potential surfaces are well described by undistorted harmonic oscillators, i.e., no change in frequency between ground and emitting states, and the transition moment is constant.
Figure 4b shows calculated PL spectra from the two-mode model using Equations (1) and (2) illustrating the effect of the low frequency C–H wag displacement on the MIME interval. Initially, displacements of both modes are set to 1.0 and spectra are calculated for large (Γ = 300 cm
−1) and small (Γ = 15 cm
−1) linewidths (black dotted traces). Decreasing the C–H wag mode displacement to 0.75 results in lower intensities and a smaller contribution to the observed interval in the broadened spectrum. In this case, the MIME increases to ~1400 cm
−1 (red solid traces) from ~1300 cm
−1. On the other hand, increasing the displacement to 1.25 shifts intensity away from the 1580 cm
−1 mode causing a smaller effective progression interval of ~1150 cm
−1 (blue solid traces). Although actual vibronic lineshapes contain more displaced modes, spectra from this simple model effectively demonstrate the impact of Franck-Condon active low frequency vibrations and, more importantly, the sensitivity of the effective vibronic interval on their displacements. It is also apparent that the frequency, displacement and damping factor govern the observed MIME interval, which can never be larger than the highest frequency mode [
25]. As the temperature decreases, individual vibronic transitions become resolved and the MIME disappears, which can be replicated here by lowering the damping factor, Γ. We show that tracking changes in the MIME interval from room temperature single molecule spectra is an effective means for understanding conformational attributes of single MEH-PPV chains without the need for more time-consuming and expensive low temperature measurements. Example low temperature (ca. 20 K) PL spectra of red and blue MEH-PPV emitters are shown in the
Supplementary Materials using a similar model albeit with an additional low frequency mode to fill in regions between the main progression interval. Here, vibronic maxima are still mostly “smeared” together but these spectra are useful for illustrating improved vibronic resolution.
Unlike previous MIME studies on concentrated samples, it is not possible to measure single molecule Raman spectra for estimating individual vibrational displacements. Instead, we use a simple effective coordinate model to fit the single molecule MIME spectra that replicates the MIME interval. Fit spectra are generated for each single molecule PL spectrum and the parameters are compared between each emitting form.
Figure 5a,b shows representative fits of blue and red forms from 0.2%
w/
w 2 MDa polystyrene as-cast and annealed samples, respectively (similar sub-ensemble fits for the 50 kDa samples are included in the
Supplementary Materials, Figures S3 and S4). Despite lower signal-to-noise ratios of single molecule spectra, the fitting procedure is able to distinguish differences in observed vibronic intervals between each form.
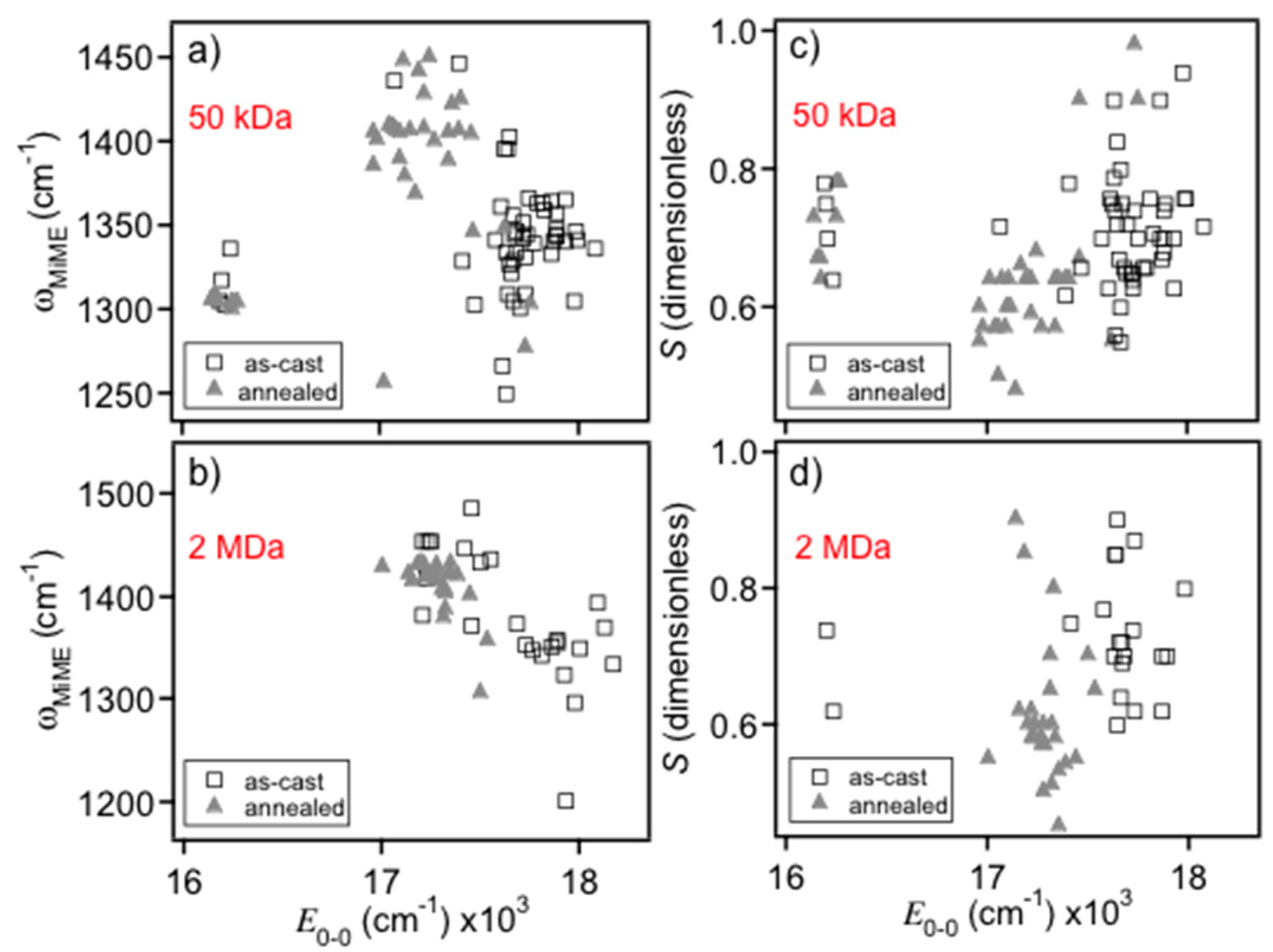
Figure 6 shows scatter plots of the MIME frequency (
Figure 6a,b) and apparent Huang–Rhys factor (
S; c,d) generated from the fits which show distinct bimodality behavior similar to
E0-0 histograms of
Figure 2 and
Figure 3. The average MIME frequency for blue-emitting MEH-PPV chains was ~1350 ± 60 cm
−1 compared to ~1430 ± 40 cm
−1 for red-emitters consistent with our previous work on solution and solid forms of MDMO-PPV. Average apparent
S values are also smaller for the red form, in agreement with recent findings that the red form behaves as a J-aggregate [
15]. It is important to stress that this parameter is used to reproduce the overall line shape profile (i.e., effective vibronic intervals) meaning that both exciton and vibronic coupling are collectively expressed in fitted
S values.
Differences in the observed MIME intervals between red and blue forms are proposed to originate chiefly from variations in displaced low frequency modes, most notably the out-of-plane C–H wag at ~966 cm
−1. Based on our previous work, this mode showed the largest change between solution and solid polymer forms, which is reasonable since larger intensities (displacements) are expected for torsionally disordered chains. As demonstrated in
Figure 4b,c, larger displacements in this mode shift intensity away from higher frequency modes resulting in a smaller observed MIME interval range of ~1220–1320 cm
−1. Displaced low frequency modes effectively “fill in” regions between vibronic peaks and, while a similar effect can be obtained by increasing the linewidth broadening parameter, Γ, the fit spectrum no longer matches with the linewidth of the 0-0 electronic origin transition. On the other hand, the larger observed MIME interval in the red emitting form implies a smaller C–H mode displacement where intensity is shifted toward the higher frequency modes (i.e., the ~1580 cm
−1 mode), as seen in
Figure 4b,c. This simple analysis of single chain PL lineshapes reveals useful insights into the conformational characteristics of each emitting form far beyond simply categorizing emission energies. Namely, the lower progression interval observed in the blue form indicates larger displacements of the out-of-plane C–H wag and, hence, larger torsional distortions of the backbone conformation. This feature is also consistent with the apparent shorter conjugation lengths of the chromophore segments (i.e., torsional induced exciton localization). Dispersing single molecules in a heavy matrix or applying SVA treatments, results in planarization of chain segments due to swelling of the matrix thus allowing chains to self-fold into more ordered, rod-like or collapsed structures [
21,
22,
23,
24]. The increase of chain planarity lowers the C–H wag displacement resulting in a larger MIME interval as seen in PL line shapes of the red MEH-PPV form. Overall, interpreting spectral changes in terms of the MIME model yields useful insights into the conformational characteristics of the polymer backbone segments as well as the structural factors that change upon conversion between these different emitting forms.
It is also necessary to consider the possibility that the difference in MEH-PPV single chain line shapes between different emitting forms arises from multiple electronic origins (emitting chromophores). For example, Basché and coworkers reported single chain MEH-PPV spectra dispersed in different host matrices displaying multiple origins at low temperatures (ca. 1.2 K) [
34]. These transitions are typically spaced close enough so that linewidth broadening at higher temperatures should cause coalescence into an effective progression interval. We found comparatively few spectra displaying evidence of multiple emitters that were excluded from the analysis mainly because these line shapes tend to show spectral diffusion due to photo-oxidation of individual chromophores of different energies and energy funnels [
9,
35]. Since spectral diffusion was small and virtually no spectra showed switching between blue or red emitters, we conclude that multiple origins are not responsible for differences in MIME intervals. We also used lower excitation intensities (i.e., 100 W/cm
2 vs. 2 kW/cm
2) thus lowering photo-oxidation yields and providing better photo-stability and small spectral diffusion. This is especially important for tracking the MIME phenomenon in single molecules because “off” periods limit overall signal-to-noise ratios making it difficult to reliably assess the MIME phenomenon.
3.2. Ultra-Red MEH-PPV Emitters
We now return to the appearance of ultra-red emitters for MEH-PPV chains dispersed in the lower MW polytstyrene matrix. These minority emitters become most apparent after annealing and the MIME progression interval in this species is closer to the blue form (~1330 cm
−1). The latter feature indicates a larger contribution from lower frequency vibrations (i.e., out-of-plane C–H wag) and lower intrachain chromophore order. However,
E0-0 values and good photostability (i.e., absence of flickering in transients) of the ultra-red form are similar to PL spectra of MEH-PPV aggregates. For example, MEH-PPV nanoparticles can be fabricated by rapidly injecting well-dissolved chains into a non-solvent, such as water. PL spectra of these nanoparticle aggregates and the ultra-red form are included in the
Supplementary Materials that show very similar line shape characteristics. It has also been demonstrated that these larger particles exhibit no flickering behavior and are typically photostable for long times [
36,
37]. We conclude that ultra-red emitters are most likely small aggregates of MEH-PPV chains that associated upon SVA treatment.
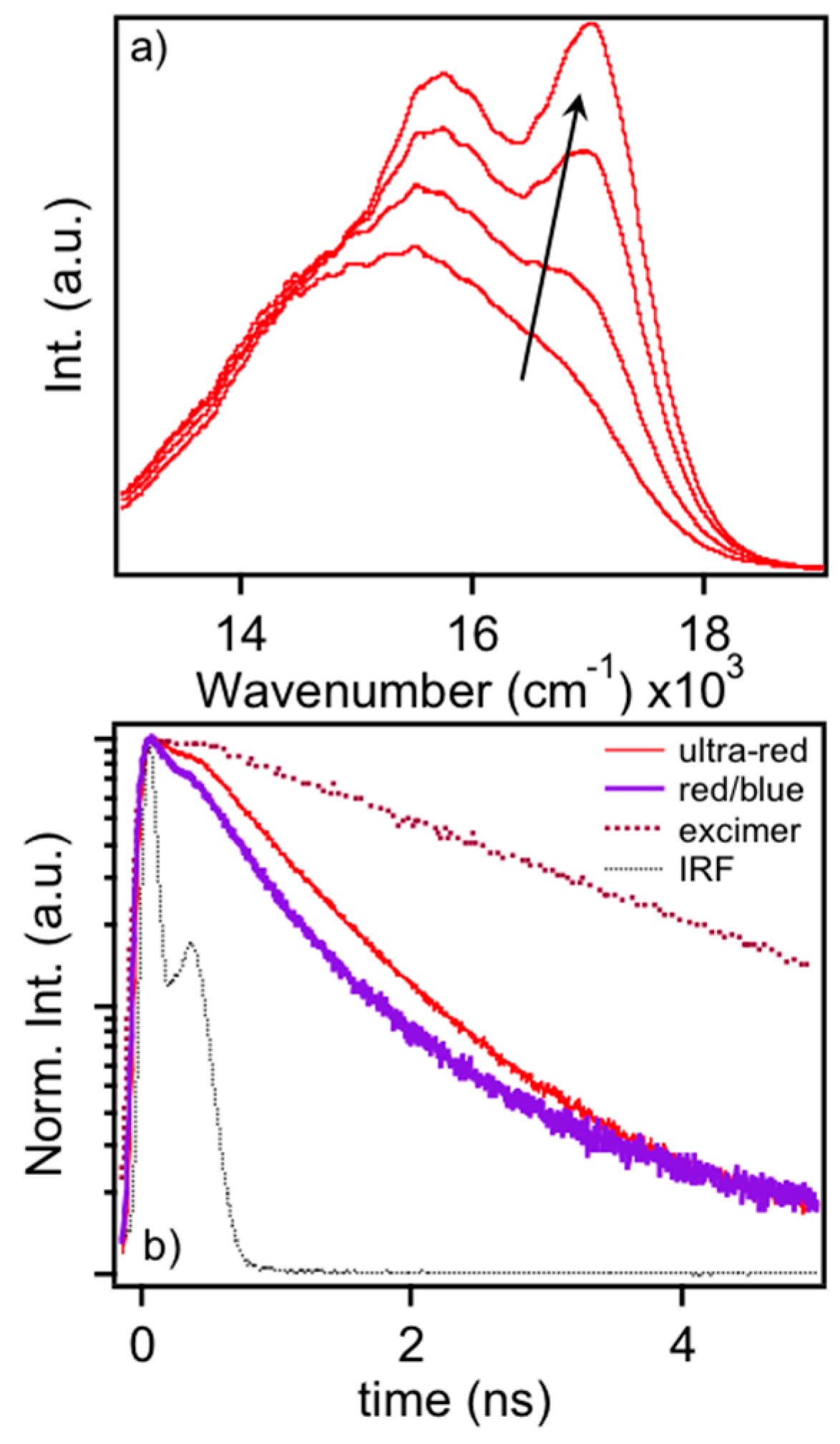
The similarity of ultra-red MEH-PPV emitters to aggregated or agglomerated MEH-PPV nanoparticles illustrates the rapid convergence of single molecule emitters to the bulk-like PL emission from thin films. Here, rapid energy funneling to minority emissive traps leads to their red-shifted PL line shapes relative to dilute solution phase PL line shape characteristics. Interestingly, application of thermal annealing to MEH-PPV thin films reveals further collapse of this line shape to one resembling an excimer type state, as shown in
Figure 7a. Subsequent SVA treatment restores PL line shapes to the characteristic form as seen in aggregates or the ultra-red variant. In the excimer form, there are a greater number of vibrational modes distorted since the excitation is intermolecular in nature. Restoration of the typical PL line shape in the condensed phase indicates these excitons possess greater intramolecular character and, consequently, are more sensitive to chromophore structure and chain conformation.
To further sort out the structural differences as well as those of excitons between typical red/blue forms and the bulk-like ultra-red and excimer forms of MEH-PPV, we use time-correlated single photon counting techniques to study the PL decay dynamics in each form.
Figure 7b shows ensemble averaged PL decays of single MEH-PPV molecules. Corresponding PL spectra were measured to correlate the specific emitting form to decay characteristics. We found very little differences between the single molecule blue and red PL forms and average these decays together in
Figure 7b, which show decay times of ~300 ps similar to previous work [
13]. Average decay times for the ultra-red species were found to be ~500 ps whereas the excimer-like transition in thermally annealed bulk films is ~2 ns. In addition to the increase in PL decay time, the high photo-stability of the ultra-red species indicates multi-chromophoric behavior where virtually no flickering or bleaching occurs within the timeframe of the measurement (>3 min). Combined with PL spectral characteristics, these data clearly show stark differences between the intra- and inter-molecular exciton regimes.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






