

Predicting the Postmortem Interval Based on Gravesoil Microbiome Data and a Random Forest Model

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design and Sample Collection
2.2. Soil Physical and Chemical Properties
2.3. DNA Extraction, PCR Amplification and Sequencing
2.4. Data Analysis
3. Results
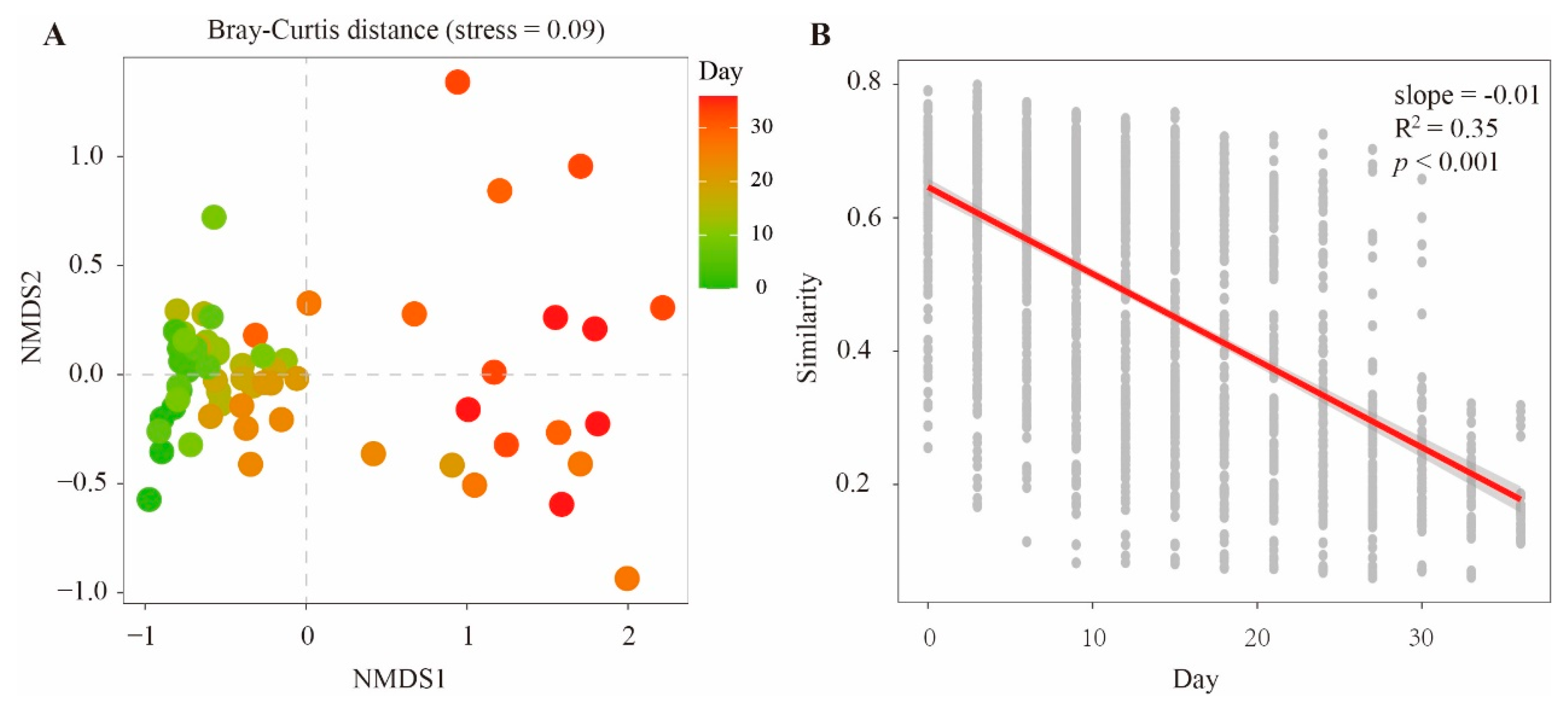
3.1. Sequencing and Bacterial Community Composition during Decomposition
3.2. Bacterial Succession Pattern during Cadaver Decomposition
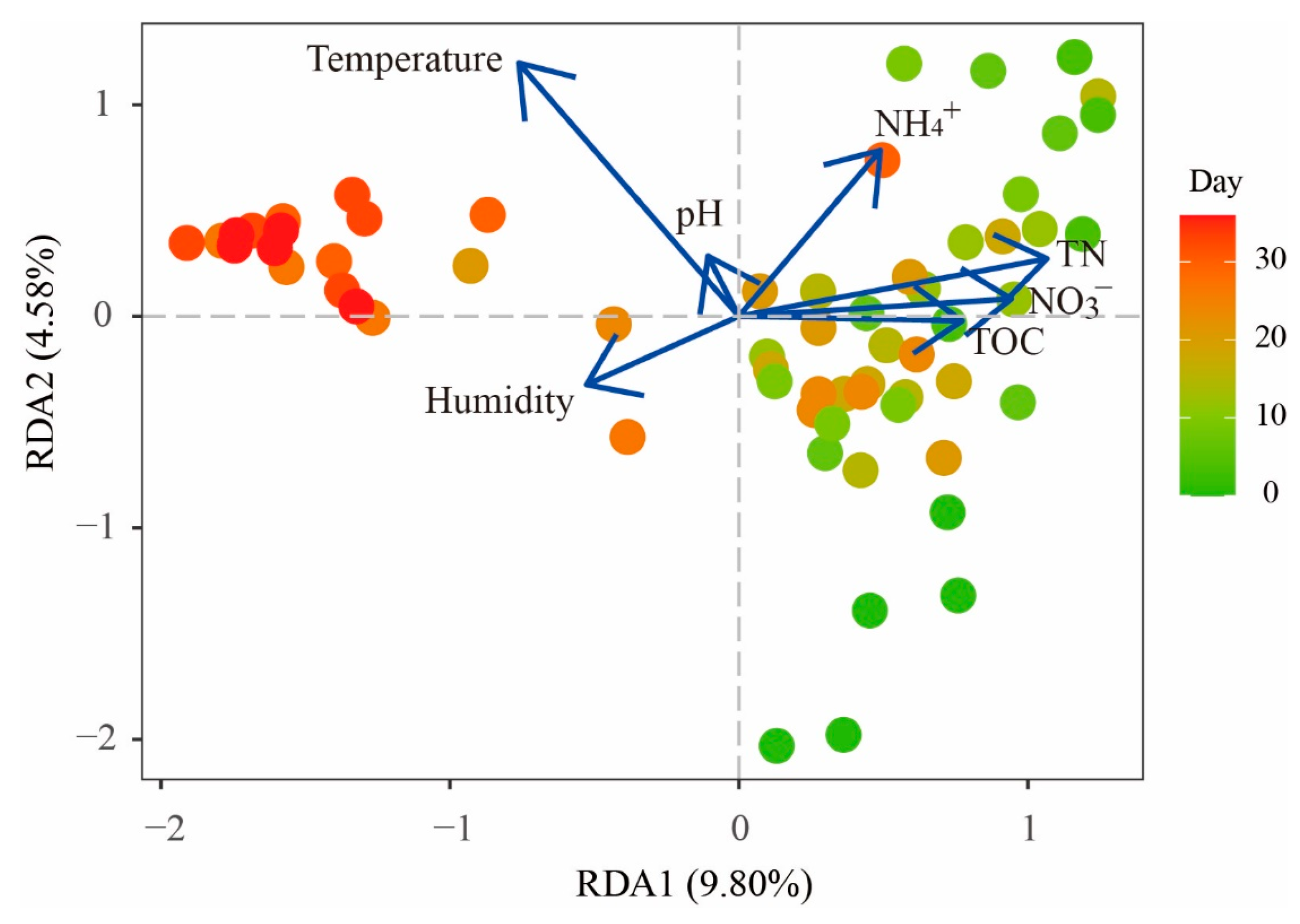
3.3. Effect of Environmental Factors on the Bacterial Community Composition
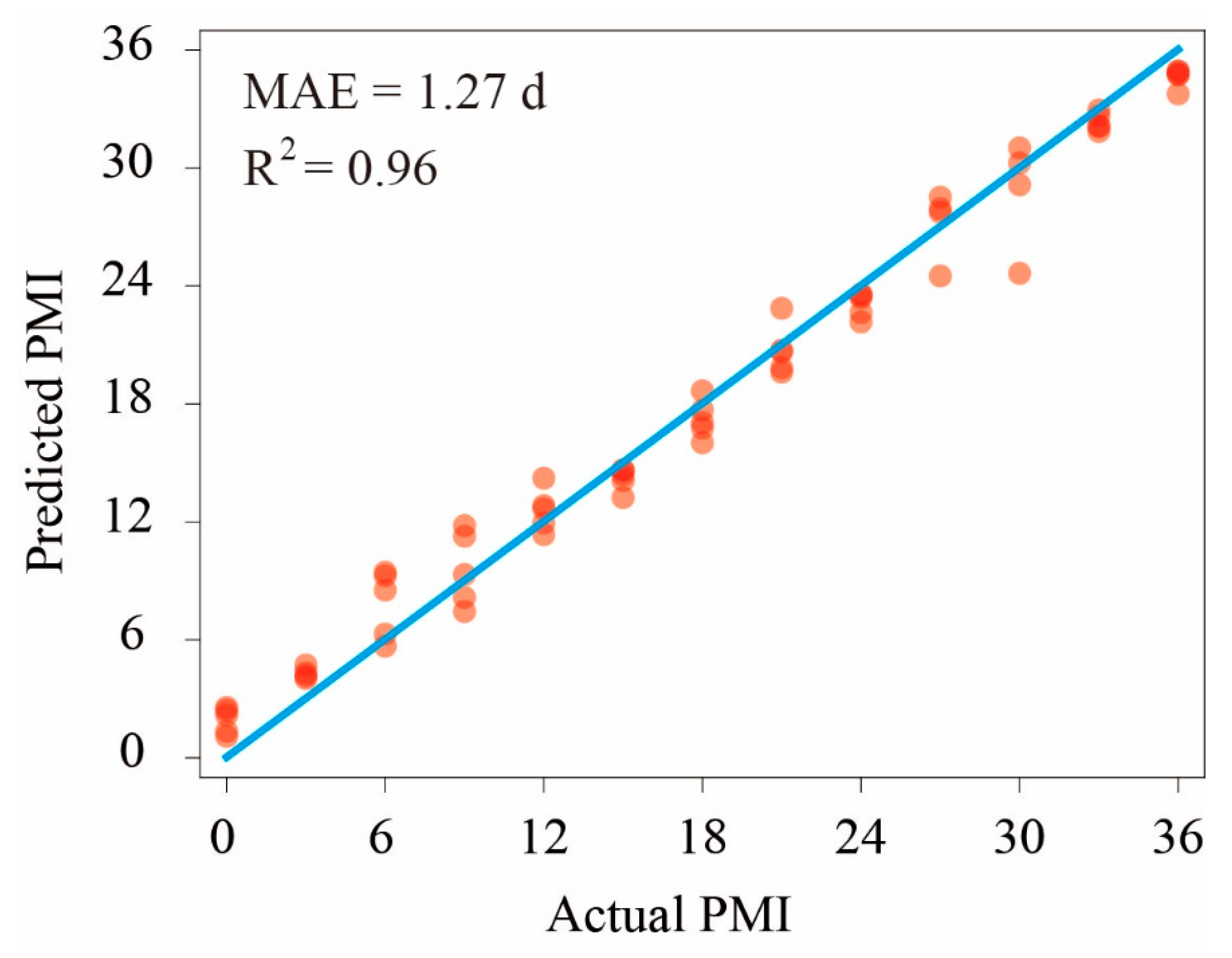
3.4. Bacterial Taxonomic Biomarkers for the PMI Determined Using the RF Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Ethical Approval
Data Availability Statement
Conflicts of Interest
References
- Saks, M.J.; Koehler, J.J. The coming paradigm shift in forensic identification science. Science 2005, 309, 892–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metcalf, J.L. Estimating the postmortem interval using microbes: Knowledge gaps and a path to technology adoption. Forensic Sci. Int.-Gen. 2019, 38, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henßge, C.; Madea, B. Estimation of the time since death in the early post-mortem period. Forensic Sci. Int. 1996, 144, 167–175. [Google Scholar] [CrossRef]
- Wyler, D.; Marty, W.; Bar, W. Correlation between the post-mortem cell content of cerebrospinal fluid and time of death. Int. J. Legal Med. 1994, 106, 194–199. [Google Scholar] [CrossRef]
- Mikami, H.; Terazawa, K.; Takatori, T.; Tokudome, S.; Tsukamoto, T.; Haga, K. Estimation of time of death by quantification of melatonin in corpses. Int. J. Legal Med. 1994, 107, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Pittner, S.; Monticelli, F.C.; Pfisterer, A.; Zissler, A.; Sänger, A.M.; Stoiber, W.; Steinbacher, P. Postmortem degradation of skeletal muscle proteins: A novel approach to determine the time since death. Int. J. Legal Med. 2016, 130, 421–431. [Google Scholar] [CrossRef]
- Young, S.T.; Wells, J.D.; Hobbs, G.R.; Bishop, C.P. Estimating postmortem interval using RNA degradation and morphological changes in tooth pulp. Forensic Sci. Int. 2013, 229, e1–e163. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Lesnikova, I.; Funder, A.M.; Banner, J. DNA and RNA analysis of blood and muscle from bodies with variable postmortem intervals. Forensic Sci. Med. Pathol. 2014, 10, 322–328. [Google Scholar] [CrossRef]
- Amendt, J.; Campobasso, C.P.; Gaudry, E.; Reiter, C.; LeBlanc, H.N.; Hall, M.J.R. Best practice in forensic entomology—Standards and guidelines. Int. J. Legal Med. 2007, 121, 90–104. [Google Scholar] [CrossRef]
- Guo, Y.; Cai, J.; Chang, Y.; Li, X.; Liu, Q.; Wang, X.; Wang, X.; Zhong, M.; Wen, J.; Wang, J. Identification of forensically important sarcophagid flies (Diptera: Sarcophagidae) in China, based on COI and 16S rDNA gene sequences. J. Forensic Sci. 2011, 56, 1534–1540. [Google Scholar] [CrossRef]
- Zhang, J.; Li, B.; Wang, Q.; Wei, X.; Feng, W.; Chen, Y.; Huang, P.; Wang, Z. Application of fourier transform infrared spectroscopy with chemometrics on postmortem interval estimation based on pericardial fluids. Sci. Rep. 2017, 7, 18013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metcalf, J.L.; Parfrey, L.W.; Gonzalez, A.; Lauber, C.L.; Knight, D.; Ackermann, G.; Humohrey, G.C.; Gebert, M.J.; van Treuren, W.; Berg-Lyons, D.; et al. A microbial clock provides an accurate estimate of the postmortem interval in a mouse model system. eLife 2013, 2, e01104. [Google Scholar] [CrossRef] [PubMed]
- Pittner, S.; Bugelli, V.; Benbow, M.E.; Ehrenfellner, B.; Zissler, A.; Campobasso, C.P.; Oostra, R.-J.; Aalders, M.C.G.; Zehner, R.; Lutz, L.; et al. The applicability of forensic time since death estimation methods for buried bodies in advanced decomposition stages. PLoS ONE 2020, 15, e0243395. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.M.; Pasternak, Z.; Mason, C.E.; Elhaik, E. Forensic application of microbiomics: A review. Front. Microbiol. 2021, 11, 608101. [Google Scholar] [CrossRef] [PubMed]
- Phan, K.; Barash, M.; Spindler, X.; Gunn, P.; Roux, C. Retrieving forensic information about the donor through bacterial profiling. Int. J. Legal Med. 2020, 134, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Lienhard, P.; Tivet, F.; Chabanne, A.; Dequiedt, S.; Lelièvre, M.; Sayphoummie, S.; Leudphanane, B.; Prévost-Bouré, N.C.; Séguy, L.; Maron, P.-A.; et al. No-till and cover crops shift soil microbial abundance and diversity in Laos tropical grasslands. Agron. Sustain. Dev. 2013, 33, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Burcham, Z.M.; Pechal, J.L.; Schmidt, C.J.; Bose, J.L.; Rosch, J.W.; Benbow, M.E.; Jordan, H.R. Bacterial community succession, transmigration, and differential gene transcription in a controlled vertebrate decomposition. Front. Microbiol. 2019, 10, 745. [Google Scholar] [CrossRef]
- Pechal, J.L.; Crippen, T.L.; Benbow, M.E.; Tarone, A.M.; Dowd, S.; Tomberlin, J.K. The potential use of bacterial community succession in forensics as described by high throughput metagenomic sequencing. Int. J. Legal Med. 2014, 128, 193–205. [Google Scholar] [CrossRef]
- Carter, D.O.; Metcalf, J.L.; Bibat, A.; Knight, R. Seasonal variation of postmortem microbial communities. Forensic Sci. Med. Pathol. 2015, 11, 202–207. [Google Scholar] [CrossRef]
- Johnson, H.R.; Trinidad, D.D.; Guzman, S.; Khan, Z.; Parziale, J.V.; DeBruyn, J.M.; Lents, N.H. A machine learning approach for using the postmortem skin microbiome to estimate the posymortem interval. PLoS ONE 2016, 11, e0167370. [Google Scholar] [CrossRef]
- Belk, A.; Xu, Z.Z.; Carter, D.O.; Lynne, A.; Bucheli, S.; Knight, R.; Metcalf, J.L. Microbiome data accurately predicts the postmortem interval using random forest regression models. Genes 2018, 9, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBruyn, J.M.; Hauther, K.A. Postmortem succession of gut microbial communities in deceased human subjects. PeerJ 2017, 5, e3437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Wang, Q.; Zhang, K.; Wu, H.; Wang, G.; Cai, W.; Yu, K.; Sun, Q.; Fan, S.; Wang, Z. Analysis of postmortem intestinal microbiota successional patterns with application in postmortem interval estimation. Microb. Ecol. 2021, 84, 1087–1102. [Google Scholar] [CrossRef] [PubMed]
- Adserias-Garriga, J.; Quijada, N.M.; Hernandez, M.; Lázaro, D.R.; Steadman, D.; Garcia-Gil, L.J. Dynamics of the oral microbiota as a tool to estimate time since death. Mol. Oral Microbiol. 2017, 32, 511–516. [Google Scholar] [CrossRef]
- Dong, K.; Xin, Y.; Cao, F.; Huang, Z.; Sun, J.; Peng, M.; Liu, W.; Shi, P. Succession of oral microbiota community as a tool to estimate postmortem interval. Sci. Rep. 2019, 9, 13063. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, M.; Qi, X.; Shi, L.; Zhang, J.; Zhang, X.; Yang, T.; Ren, J.; Liu, F.; Zhang, G.; et al. Predicting the postmortem interval of burial cadavers based on microbial community succession. Forensic Sci. Int. Gen. 2021, 52, 102488. [Google Scholar] [CrossRef]
- Damann, F.E.; Williams, D.E.; Layton, A.C. Potential use of bacterial community succession in decaying human bone for estimating postmortem interval. J. Forensic Sci. 2015, 60, 844–850. [Google Scholar] [CrossRef]
- Carter, D.O.; Yellowlees, D.; Tibbett, M. Temperature affects microbial decomposition of cadavers (Rattus rattus) in constrasting soils. Appl. Soil Ecol. 2008, 40, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Giles, S.B.; Harrison, K.; Errickson, D.; Márquez-Grant, N. The effect of seasonality on the application of accumulated degree-days to estimate the early post-mortem interval. Forensic Sci. Int. 2020, 315, 110419. [Google Scholar] [CrossRef]
- Carter, D.O.; Yellowlees, D.; Tibbett, M. Moisture can be the dominant environmental parameter governing cadaver decomposition in soil. Forensic Sci. Int. 2010, 200, 60–66. [Google Scholar] [CrossRef]
- Huang, Z.; Zhao, F.; Wang, M.; Qi, K.; Wu, J.; Zhang, S. Soil chemical properties and geographical distance exerted effects on arbuscular mycorrhizal fungal community composition in pear orchards in Jiangsu Province, China. Appl. Soil Ecol. 2019, 142, 18–24. [Google Scholar] [CrossRef]
- Bao, S.D. Agricultural Chemical Analysis of Soil; China Agriculture Press: Beijing, China, 2005. [Google Scholar]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gu, Y.; Shen, M.; Li, H.; Zhang, K.; Wang, Q.; Wei, X.; Zhang, H.; Wu, D.; Yu, K.; et al. Predicting postmortem interval based on microbial community sequences and machine learning algorithms. Environ. Microbiol. 2020, 22, 2273–2291. [Google Scholar] [CrossRef] [PubMed]
- Hyun, C.; Kim, H.; Ryu, S.; Kim, W. Preliminary study on microeukaryotic community analysis using NGS technology to determine postmortem submersion interval (PMSI) in the drowned pig. J. Microbiol. 2019, 57, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, J.L.; Xu, Z.Z.; Weiss, S.; Lax, S.; van Treuren, W.; Hyde, E.R.; Sing, S.J.; Amir, A.; Larsen, P.; Sangwan, N.; et al. Microbial community assembly and metabolic function during mammalian corpse decomposition. Science 2016, 351, 158–162. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Minick, K.J.; Strickland, M.S.; Wicking, K.G.; Crippen, T.L.; Tarone, A.M.; Benbow, M.E.; Sufrin, N.; Tomberlin, J.K.; Pechal, J.L. Temporal and spatial impact of human cadaver decomposition on soil bacterial and arthropod community structure and function. Front. Microbiol. 2018, 8, 2616. [Google Scholar] [CrossRef]
- Zhu, K.; Wang, Q.; Zhang, Y.; Zarif, N.; Ma, S.; Xu, L. Variation in soil bacterial and fungal community composition at different successional stages of a broad-leaved Korean pine forest in the Lesser Hinggan Mountains. Forest 2022, 13, 625. [Google Scholar] [CrossRef]
- Neufeld, J.D.; Mohn, W.W. Unexpectedly high bacterial diversity in arctic tundra relative to boreal forest soils, revealed by serial analysis of ribosomal sequence tags. Appl. Environ. Microbial. 2005, 71, 5710–5718. [Google Scholar] [CrossRef] [Green Version]
- Curtis, P.; Nakatsu, C.H.; Konopka, A. Aciduric proteobacteria isolated from pH 2.9 soil. Arch. Microbiol. 2002, 178, 65–70. [Google Scholar] [CrossRef]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Spain, A.M.; Krumholz, L.R.; Elshahed, M.S. Abundance, composition, diversity and novelty of soil Proteobacteria. ISME J. 2009, 3, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, J.; Yang, F.; E, Y.; Raza, W.; Huang, Q.; Shen, Q. Application of bioorganic fertilizer significantly increased apple yield and shaped bacterial community structure in orchard soil. Microb. Ecol. 2017, 73, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Wasi, S.; Tabrez, S.; Ahmad, M. Use of Pseudomonas spp. for the bioremediation of environmental pollutants: A review. Environ. Monit. Assess. 2013, 185, 8147–8155. [Google Scholar] [CrossRef]
- Hol, W.H.G.; Bezemer, M.; Biere, A. Getting the ecology into interactions between plants and the plant growth-promoting bacterium Psedomonas fluorescens. Front. Plant Sci. 2013, 4, 81. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wu, P.; Hao, B.; Yu, Z. Heterotrophic nitrification and aerobic denitrification by the bacterium Pseudomonas stutzeri YZN-001. Bioresour. Technol. 2011, 102, 9866–9869. [Google Scholar] [CrossRef]
- Zhang, X.; Xia, Y.; Zeng, Y.; Sun, X.; Tao, R.; Mei, Y.; Qu, M. Simultaneous nitrification and denitrification by Pseudomonas sp. Y-5 in a high nitrogen environment. Environ. Sci. Pollut. Res. 2022, 29, 69491–69501. [Google Scholar] [CrossRef]
- Navarrete, A.A.; Kuramae, E.E.; de Hollander, M.; Pijl, A.S.; van Veen, J.A.; Tsai, S.M. Acidobacterial community responses to agricultural management of soybean in Amazon forest soils. FEMS Microbiol. Ecol. 2013, 83, 607–621. [Google Scholar] [CrossRef] [Green Version]
- Foesel, B.U.; Nägele, V.; Naether, A.; Wüst, P.K.; Weinert, J.; Bonkowski, M.; Lohaus, G.; Polle, A.; Alt, F.; Oelmann, Y.; et al. Determinants of Acidobacteria activity inferred from the relative abundances of 16S rRNA transcripts in German grassland and forest soils. Environ. Microbiol. 2014, 16, 658–675. [Google Scholar] [CrossRef]
- Barns, S.M.; Takala, S.L.; Kuske, C.R. Wide distribution and diversity of members of the bacterial kingdom Acidobacterium in the environment. Appl. Environ. Microbiol. 1999, 65, 1731–1737. [Google Scholar] [CrossRef]
- Kielak, A.M.; Barreto, C.; Kowalchuk, G.A.; van Veen, J.A.; Kuramae, E.E. The ecology of Acidobacteria: Moving beyond genes and genomes. Front. Microbiol. 2016, 7, 744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, O.C.; Yang, X.; Fu, Y.; Feng, Z.; Sha, L.; Casper, P.; Zou, X. 16S rRNA gene analyses of bacterial community structures in the soils of evergreen broad-leaved forests in south-west China. FEMS Microbiol. Ecol. 2006, 58, 247–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Juottonen, H.; Siivonen, P.; Quesada, C.L.; Tuomi, P.; Pulkkinen, P.; Yrjälä, K. Spatial patterns of microbial diversity and activity in an aged creosote-contaminated site. ISME J. 2014, 8, 2131–2142. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Dong, J.; Ding, D.; Long, L.; Suo, A.; Lin, X.; Yang, Q.; Lin, L.; Zhang, Y.; Ling, J. Rhizosphere microbiome dynamics in tropical seagrass under short-term inorganic nitrogen fertilization. Environ. Sci. Pollut. Res. 2021, 28, 19021–19033. [Google Scholar] [CrossRef]
- Liu, C.; Dong, Y.; Hou, L.; Deng, N.; Jiao, R. Acidobacteria community responses to nitrogen dose and form in Chinese fir plantations in southern China. Curr. Microbiol. 2017, 74, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Lee, S.; Hallam, S.J.; Mohn, W.W. Bacterial, archaeal and eukaryal community structures throughout soil horizons of harvested and naturally disturbed forest stands. Environ. Microbiol. 2009, 11, 3045–3062. [Google Scholar] [CrossRef] [PubMed]
- Ryckeboer, J.; Mergaert, J.; Coosemans, J.; Deprins, K.; Swings, J. Microbiological aspects of biowaste during composting in a monitored compost bin. J. Appl. Microbiol. 2003, 94, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Morrissey, E.M.; Mau, R.L.; Schwartz, E.; McHugh, T.A.; Dijkstra, P.; Koch, B.J.; Marks, J.C.; Hungate, B.A. Bacterial carbon use plasticity, phylogenetic diversity and the priming of soil organic matter. ISME J. 2017, 11, 1890–1899. [Google Scholar] [CrossRef]
- Fierer, N.; Lauber, C.L.; Ramirez, K.S.; Zaneveld, J.; Bradford, M.A.; Knight, R. Comparative metagenomic phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2012, 6, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Eo, J.; Park, K.-C. Long-term effects of imbalanced fertilization on the composition and diversity of soil bacterial community. Agric. Ecosyst. Environ. 2016, 231, 176–182. [Google Scholar] [CrossRef]
- Procopio, N.; Ghignone, S.; Williams, A.; Chamberlain, A.; Mello, A.; Buckley, M. Metabarcoding to investigate changes in soil microbial communities within forensic burial contexts. Forensic Sci. Int.-Gen. 2019, 39, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, P.; van Zwieten, L.; Tu, J.; Gan, W.; Lu, S.; Wang, H.; Wu, L. Edaphic variables influence soil bacterial structure under successive fertilization of Paulownia plantation substituting native vegetation. J. Soil. Sediment. 2021, 21, 2922–2937. [Google Scholar] [CrossRef]
- Olakanye, A.O.; Ralebitso-Senior, T.K. Soil metabarcoding identifies season indicators and differentiators of pig and Agrostis/Festuca spp. decomposition. Forensic Sci. Int. 2018, 288, 53–58. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RDA1 | RDA2 | r2 | p Value | |
|---|---|---|---|---|
| pH | −0.130 | 0.991 | 0.018 | 0.56 |
| TC | 0.996 | 0.085 | 0.060 | 0.174 |
| TN | 0.899 | 0.438 | 0.143 | 0.012 |
| NH4+ | 0.457 | 0.890 | 0.177 | 0.003 |
| NO3− | 0.969 | 0.246 | 0.095 | 0.05 |
| Temperature | −0.303 | 0.953 | 0.345 | 0.001 |
| Humidity | −0.729 | −0.685 | 0.058 | 0.172 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, C.; Song, Y.; Mao, D.; Cao, Y.; Qiu, B.; Gui, P.; Wang, H.; Zhao, X.; Huang, Z.; Sun, L.; et al. Predicting the Postmortem Interval Based on Gravesoil Microbiome Data and a Random Forest Model. Microorganisms 2023, 11, 56. https://doi.org/10.3390/microorganisms11010056
Cui C, Song Y, Mao D, Cao Y, Qiu B, Gui P, Wang H, Zhao X, Huang Z, Sun L, et al. Predicting the Postmortem Interval Based on Gravesoil Microbiome Data and a Random Forest Model. Microorganisms. 2023; 11(1):56. https://doi.org/10.3390/microorganisms11010056
Chicago/Turabian StyleCui, Chunhong, Yang Song, Dongmei Mao, Yajun Cao, Bowen Qiu, Peng Gui, Hui Wang, Xingchun Zhao, Zhi Huang, Liqiong Sun, and et al. 2023. "Predicting the Postmortem Interval Based on Gravesoil Microbiome Data and a Random Forest Model" Microorganisms 11, no. 1: 56. https://doi.org/10.3390/microorganisms11010056
APA StyleCui, C., Song, Y., Mao, D., Cao, Y., Qiu, B., Gui, P., Wang, H., Zhao, X., Huang, Z., Sun, L., & Zhong, Z. (2023). Predicting the Postmortem Interval Based on Gravesoil Microbiome Data and a Random Forest Model. Microorganisms, 11(1), 56. https://doi.org/10.3390/microorganisms11010056