
Gut Microbiome Studies in Livestock: Achievements, Challenges, and Perspectives

 , and
, and 
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Next-Generation Sequencing Techniques
2.1. Amplicon Metabarcoding
2.2. Shotgun Sequencing
2.3. Metatranscriptomics
3. The Significance of Microbiome Studies in Livestock Species
4. Microbiome Studies in Livestock Species
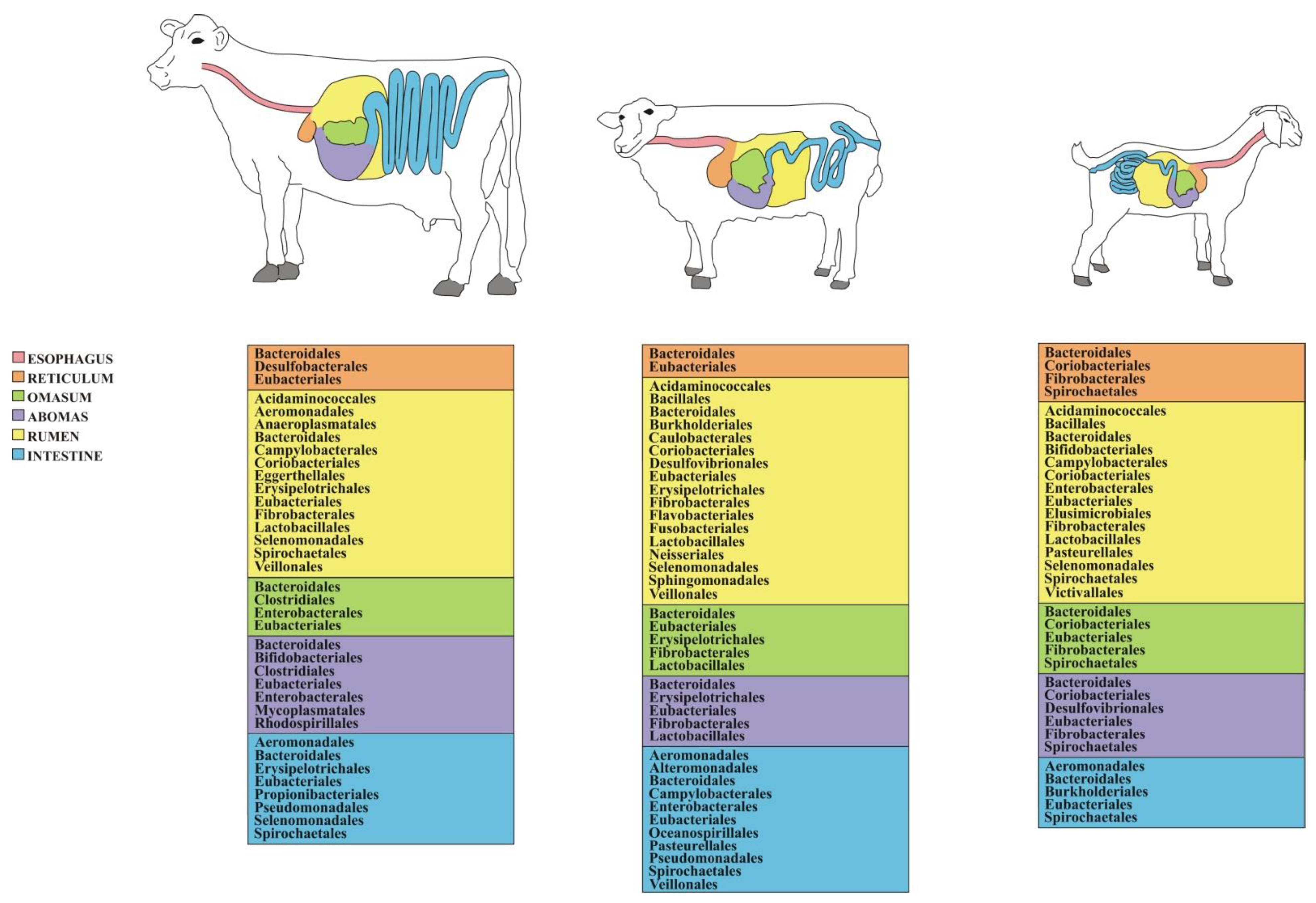
4.1. Ruminants
4.1.1. Cattle
4.1.2. Cattle Microbiome Profiling
4.1.3. Sheep
4.1.4. Sheep Microbiome Profiling
4.1.5. Goat
4.1.6. Goat Microbiome Profiling
4.2. Monogastric
4.2.1. Pig
4.2.2. Pig Microbiome Profiling
4.2.3. Chicken
4.2.4. Chicken Microbiome Profiling
5. Resistome
6. Metagenome and Functional Profile Prediction
7. Gut Microbiome, Health, and Welfare in Livestock
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amos, G.C.A.; Logan, A.; Anwar, S.; Fritzsche, M.; Mate, R.; Bleazard, T.; Rijpkema, S. Developing standards for the microbiome field. Microbiome 2020, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- European OneHealth/EcoHealth Workshop. Available online: http://www.biodiversity.be/health/58 (accessed on 20 May 2022).
- Park, W. Gut microbiomes and their metabolites shape human and animal health. J. Microbiol. 2018, 56, 151–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, G.O.; Gruninger, R.J.; Badhan, A.; McAllister, T.A. Mining the rumen for fibrolytic feed enzymes. Anim. Front. 2016, 6, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Weimer, P.J. Redundancy, resilience, and host specificity of the ruminal microbiota: Implications for engineering improved ruminal fermentations. Front. Microbiol. 2015, 6, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malmuthuge, N.; Guan, L.L. Gut microbiome and omics: A new definition to ruminant production and health. Anim. Front. 2016, 6, 8–12. [Google Scholar] [CrossRef]
- Alexander, T.W.; Plaizier, J.C. From the Editors: The importance of microbiota in ruminant production. Anim. Front. 2016, 6, 4–7. [Google Scholar] [CrossRef] [Green Version]
- Cholewinska, P.; Czyz, K.; Nowakowski, P.; Wyrostek, A. The microbiome of the digestive system of ruminants—A review. Anim. Health Res. Rev. 2020, 21, 3–14. [Google Scholar] [CrossRef]
- Alexandratos, N.; Bruinsma, J. World Agriculture towards 2030/2050: The 2012 Revision; ESA Working Paper 12-03; FAO: Rome, Italy, 2012. [Google Scholar]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Verges, M.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics—A guide from sampling to data analysis. Microb. Inform. Exp. 2012, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- National Human Genome Research Institute. Metagenomics. Available online: https://www.genome.gov/genetics-glossary/Metagenomics (accessed on 12 June 2022).
- Wareth, G.; El-Diasty, M.; Abdel-Hamid, N.H.; Holzer, K.; Hamdy, M.E.R.; Moustafa, S.; Shahein, M.A.; Melzer, F.; Beyer, W.; Pletz, M.W.; et al. Molecular characterization and antimicrobial susceptibility testing of clinical and non-clinical Brucella melitensis and Brucella abortus isolates from Egypt. One Health 2021, 13, 100255. [Google Scholar] [CrossRef]
- Hristov, A.N.; Ivan, M.; Rode, L.M.; McAllister, T.A. Fermentation characteristics and ruminal ciliate protozoal populations in cattle fed medium- or high-concentrate barley-based diets. J. Anim. Sci. 2001, 79, 515–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drancourt, M.; Bollet, C.; Carlioz, A.; Martelin, R.; Gayral, J.-P.; Raoult, R. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J. Clin. Microbiol. 2000, 38, 3623–3630. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Wang, Y.; Qian, P.Y. Sensitivity and correlation of hypervariable regions in 16S rRNA genes in phylogenetic analysis. BMC Bioinform. 2016, 17, 135. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Park, J.S. Comparative analyses of the V4 and V9 regions of 18S rDNA for the extant eukaryotic community using the Illumina platform. Sci. Rep. 2020, 10, 6519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellemain, E.; Carlsen, T.; Brochmann, C.; Coissac, E.; Taberlet, P.; Kauserud, H. ITS as an environmental DNA barcode for fungi: An in silico approach reveals potential PCR biases. BMC Microbiol. 2010, 10, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [Green Version]
- Poretsky, R.; Rodriguez, R.L.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Maidak, B.L.; Olsen, G.J.; Larsen, N.; Overbeek, R.; McCaughey, M.J.; Woese, C.R. The RDP (Ribosomal Database Project). Nucleic Acids Res. 1997, 25, 109–111. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Asshauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinroth, M.D.; Belk, A.D.; Dean, C.; Noyes, N.; Dittoe, D.K.; Rothrock, M.J.; Ricke, S.C.; Myer, P.R.; Henniger, M.T.; Ramirez, G.A.; et al. Considerations and best practices in animal science 16S ribosomal RNA gene sequencing microbiome studies. J. Anim. Sci. 2022, 100, skab346. [Google Scholar] [CrossRef] [PubMed]
- National Human Genome Research Institute. Shotgun Sequencing. Available online: https://www.genome.gov/genetics-glossary/Shotgun-Sequencing (accessed on 12 June 2022).
- Sharpton, T.J. An introduction to the analysis of shotgun metagenomic data. Front. Plant. Sci. 2014, 5, 209. [Google Scholar] [CrossRef] [Green Version]
- Peabody, M.A.; Van Rossum, T.; Lo, R.; Brinkman, F.S. Evaluation of shotgun metagenomics sequence classification methods using in silico and in vitro simulated communities. BMC Bioinform. 2015, 16, 363. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- O’Malley, M.A. Metatranscriptomics. In Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013. [Google Scholar]
- Waikel, R.L.; Waikel, P.A. Metatranscriptomics. AccessScience. 2011. Available online: https://www.accessscience.com/content/article/aYB110044 (accessed on 27 November 2022).
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Bui, T.T.; Selvarajoo, K. ABioTrans: A Biostatistical Tool for Transcriptomics Analysis. Front. Genet. 2019, 10, 499. [Google Scholar] [CrossRef] [Green Version]
- FAO. How to feed the world in 2050. In Proceedings of the Expert Meeting on How to Feed the World in 2050; Food and Agriculture Organization of the United Nations: Rome, Italy, 2009. [Google Scholar]
- FAO. FAOSTAT. Available online: http://www.fao.org/faostat/en/#home (accessed on 18 March 2022).
- Pulina, G.; Milan, M.J.; Lavin, M.P.; Theodoridis, A.; Morin, E.; Capote, J.; Thomas, D.L.; Francesconi, A.H.D.; Caja, G. Invited review: Current production trends, farm structures, and economics of the dairy sheep and goat sectors. J. Dairy Sci. 2018, 101, 6715–6729. [Google Scholar] [CrossRef] [Green Version]
- Reece, W.O. Functional Anatomy and Physiology of Domestic Animals, 4th ed.; Wiley-Blackwell: Ames, IA, USA, 2009. [Google Scholar]
- Celi, P.; Cowieson, A.J.; Fru-Nji, F.; Steinert, R.E.; Kluenter, A.M.; Verlhac, V. Gastrointestinal functionality in animal nutrition and health: New opportunities for sustainable animal production. Anim. Feed Sci. Technol. 2017, 234, 88–100. [Google Scholar] [CrossRef]
- Heil, B.A.; Paccamonti, D.L.; Sones, J.L. Role for the mammalian female reproductive tract microbiome in pregnancy outcomes. Physiol. Genom. 2019, 51, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Min, B.R.; Solaiman, S.; Waldrip, H.M.; Parker, D.; Todd, R.W.; Brauer, D. Dietary mitigation of enteric methane emissions from ruminants: A review of plant tannin mitigation options. Anim. Nutr. 2020, 6, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Black, Z.; Balta, I.; Black, L.; Naughton, P.J.; Dooley, J.S.G.; Corcionivoschi, N. The Fate of Foodborne Pathogens in Manure Treated Soil. Front. Microbiol. 2021, 12, 781357. [Google Scholar] [CrossRef]
- Zalewska, M.; Blazejewska, A.; Czapko, A.; Popowska, M. Antibiotics and Antibiotic Resistance Genes in Animal Manure—Consequences of Its Application in Agriculture. Front. Microbiol. 2021, 12, 610656. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, J.; Kuehn, L.A.; Bono, J.L.; Berry, E.D.; Kalchayanand, N.; Freetly, H.C.; Benson, A.K.; Wells, J.E. Investigation of bacterial diversity in the feces of cattle fed different diets. J. Anim. Sci. 2014, 92, 683–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palumbo, F.; Squartini, A.; Barcaccia, G.; Macolino, S.; Pornaro, C.; Pindo, M.; Sturaro, E.; Ramanzin, M. A multi-kingdom metabarcoding study on cattle grazing Alpine pastures discloses intra-seasonal shifts in plant selection and faecal microbiota. Sci. Rep. 2021, 11, 889. [Google Scholar] [CrossRef]
- Mao, S.; Zhang, M.; Liu, J.; Zhu, W. Characterising the bacterial microbiota across the gastrointestinal tracts of dairy cattle: Membership and potential function. Sci. Rep. 2015, 5, 16116. [Google Scholar] [CrossRef] [Green Version]
- McGovern, E.; Kenny, D.A.; McCabe, M.S.; Fitzsimons, C.; McGee, M.; Kelly, A.K.; Waters, S.M. 16S rRNA Sequencing Reveals Relationship Between Potent Cellulolytic Genera and Feed Efficiency in the Rumen of Bulls. Front. Microbiol. 2018, 9, 1842. [Google Scholar] [CrossRef]
- Gonzalez-Recio, O.; Zubiria, I.; Garcia-Rodriguez, A.; Hurtado, A.; Atxaerandio, R. Short communication: Signs of host genetic regulation in the microbiome composition in 2 dairy breeds: Holstein and Brown Swiss. J. Dairy Sci. 2018, 101, 2285–2292. [Google Scholar] [CrossRef] [Green Version]
- Plaizier, J.C.; Li, S.; Tun, H.M.; Khafipour, E. Nutritional Models of Experimentally-Induced Subacute Ruminal Acidosis (SARA) Differ in Their Impact on Rumen and Hindgut Bacterial Communities in Dairy Cows. Front. Microbiol. 2016, 7, 2128. [Google Scholar] [CrossRef]
- Rudi, K.; Moen, B.; Sekelja, M.; Frisli, T.; Lee, M.R. An eight-year investigation of bovine livestock fecal microbiota. Vet. Microbiol. 2012, 160, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Jami, E.; Mizrahi, I. Composition and similarity of bovine rumen microbiota across individual animals. PLoS ONE 2012, 7, e33306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.W.; Connor, E.E.; Li, C.; Baldwin Vi, R.L.; Sparks, M.E. Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environ. Microbiol. 2012, 14, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Hagey, J.V.; Bhatnagar, S.; Heguy, J.M.; Karle, B.M.; Price, P.L.; Meyer, D.; Maga, E.A. Fecal Microbial Communities in a Large Representative Cohort of California Dairy Cows. Front. Microbiol. 2019, 10, 1093. [Google Scholar] [CrossRef] [PubMed]
- Durso, L.M.; Harhay, G.P.; Smith, T.P.; Bono, J.L.; Desantis, T.Z.; Harhay, D.M.; Andersen, G.L.; Keen, J.E.; Laegreid, W.W.; Clawson, M.L. Animal-to-animal variation in fecal microbial diversity among beef cattle. Appl. Environ. Microbiol. 2010, 76, 4858–4862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Global Rumen Census Collaborators; Janssen, P.H. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5, 14567. [Google Scholar] [CrossRef] [Green Version]
- Taxis, T.M.; Wolff, S.; Gregg, S.J.; Minton, N.O.; Zhang, C.; Dai, J.; Schnabel, R.D.; Taylor, J.F.; Kerley, M.S.; Pires, J.C.; et al. The players may change but the game remains: Network analyses of ruminal microbiomes suggest taxonomic differences mask functional similarity. Nucleic Acids Res. 2015, 43, 9600–9612. [Google Scholar] [CrossRef] [Green Version]
- Brulc, J.M.; Antonopoulos, D.A.; Miller, M.E.; Wilson, M.K.; Yannarell, A.C.; Dinsdale, E.A.; Edwards, R.E.; Frank, E.D.; Emerson, J.B.; Wacklin, P.; et al. Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc. Natl. Acad. Sci. USA 2009, 106, 1948–1953. [Google Scholar] [CrossRef] [Green Version]
- Wallace, R.J.; Sasson, G.; Garnsworthy, P.C.; Tapio, I.; Gregson, E.; Bani, P.; Huhtanen, P.; Bayat, A.R.; Strozzi, F.; Biscarini, F.; et al. A heritable subset of the core rumen microbiome dictates dairy cow productivity and emissions. Sci. Adv. 2019, 5, eaav8391. [Google Scholar] [CrossRef] [Green Version]
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Walker, A.W.; Roehe, R.; Watson, M. Compendium of 4941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 2019, 37, 953–961. [Google Scholar] [CrossRef]
- Xue, M.Y.; Sun, H.Z.; Wu, X.H.; Liu, J.X.; Guan, L.L. Multi-omics reveals that the rumen microbiome and its metabolome together with the host metabolome contribute to individualized dairy cow performance. Microbiome 2020, 8, 64. [Google Scholar] [CrossRef]
- Khafipour, E.; Li, S.; Plaizier, J.C.; Krause, D.O. Rumen microbiome composition determined using two nutritional models of subacute ruminal acidosis. Appl. Environ. Microbiol. 2009, 75, 7115–7124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petri, R.M.; Schwaiger, T.; Penner, G.B.; Beauchemin, K.A.; Forster, R.J.; McKinnon, J.J.; McAllister, T.A. Characterization of the Core Rumen Microbiome in Cattle during Transition from Forage to Concentrate as Well as during and after an Acidotic Challenge. PLoS ONE 2013, 8, e83424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jami, E.; Israel, A.; Kotser, A.; Mizrahi, I. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 2013, 7, 1069–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, S.; Zhang, R.; Wang, D.; Zhu, W. Impact of subacute ruminal acidosis (SARA) adaptation on rumen microbiota in dairy cattle using pyrosequencing. Anaerobe 2013, 24, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Nagata, R.; Ohkubo, A.; Ohtani, N.; Kushibiki, S.; Ichijo, T.; Sato, S. Changes in ruminal and reticular pH and bacterial communities in Holstein cattle fed a high-grain diet. BMC Vet. Res. 2018, 14, 310. [Google Scholar] [CrossRef]
- Myer, P.; Freetly, H.; Wells, J.; Smith, T.; Kuehn, L. Analysis of the gut bacterial communities in beef cattle and their association with feed intake, growth, and efficiency. J. Anim. Sci. 2017, 95, 3215–3224. [Google Scholar] [CrossRef]
- McLoughlin, S.; Spillane, C.; Claffey, N.; Smith, P.E.; O’Rourke, T.; Diskin, M.G.; Waters, S.M. Rumen Microbiome Composition Is Altered in Sheep Divergent in Feed Efficiency. Front. Microbiol. 2020, 11, 1981. [Google Scholar] [CrossRef]
- Fu, Z.; Xu, X.; Zhang, J.; Zhang, L. Effect of different feeding methods on rumen microbes in growing Chinese Tan sheep. Rev. Bras. Zootec. 2020, 49, e20190258. [Google Scholar] [CrossRef]
- Yu, S.; Zhang, G.; Liu, Z.; Wu, P.; Yu, Z.; Wang, J. Repeated inoculation with fresh rumen fluid before or during weaning modulates the microbiota composition and co-occurrence of the rumen and colon of lambs. BMC Microbiol. 2020, 20, 29. [Google Scholar] [CrossRef]
- Wang, J.; Fan, H.; Han, Y.; Zhao, J.; Zhou, Z. Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis. Asian-Australas. J. Anim. Sci. 2017, 30, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, L.; Liu, H.; Xu, T.; Zhao, N.; Zhang, X.; Geng, Y.; Kang, S.; Xu, S. Characterization of the bacterial microbiota across the different intestinal segments of the Qinghai semi-fine wool sheep on the Qinghai-Tibetan Plateau. Anim. Biosci. 2021, 34, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.; Wills, J.; Su, X.; Hewitt, R.E.; Robertson, J.; Scotti, R.; Price, D.R.G.; Bartley, Y.; McNeilly, T.N.; Krause, L.; et al. Infection with the sheep gastrointestinal nematode Teladorsagia circumcincta increases luminal pathobionts. Microbiome 2020, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Shabana, I.I.; Albakri, N.N.; Bouqellah, N.A. Metagenomic investigation of faecal microbiota in sheep and goats of the same ages. J. Taibah Univ. Sci. 2020, 15, 1–9. [Google Scholar] [CrossRef]
- Chang, J.; Yao, X.; Zuo, C.; Qi, Y.; Chen, D.; Ma, W. The gut bacterial diversity of sheep associated with different breeds in Qinghai province. BMC Vet. Res. 2020, 16, 254. [Google Scholar] [CrossRef]
- Minozzi, G.; Biscarini, F.; Dalla Costa, E.; Chincarini, M.; Ferri, N.; Palestrini, C.; Minero, M.; Mazzola, S.; Piccinini, R.; Vignola, G.; et al. Analysis of Hindgut Microbiome of Sheep and Effect of Different Husbandry Conditions. Animals 2020, 11, 4. [Google Scholar] [CrossRef]
- Zhang, Y.K.; Zhang, X.X.; Li, F.D.; Li, C.; Li, G.Z.; Zhang, D.Y.; Song, Q.Z.; Li, X.L.; Zhao, Y.; Wang, W.M. Characterization of the rumen microbiota and its relationship with residual feed intake in sheep. Animal 2021, 15, 100161. [Google Scholar] [CrossRef]
- Zeng, Y.; Zeng, D.; Ni, X.; Zhu, H.; Jian, P.; Zhou, Y.; Xu, S.; Lin, Y.; Li, Y.; Yin, Z.; et al. Microbial community compositions in the gastrointestinal tract of Chinese Mongolian sheep using Illumina MiSeq sequencing revealed high microbial diversity. AMB Express 2017, 7, 75. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.; Jin, W.; Si, H.; Yuan, Y.; Tao, Y.; Liu, J.; Wang, X.; Yang, C.; Li, Q.; Yan, X.; et al. An integrated gene catalog and over 10,000 metagenome-assembled genomes from the gastrointestinal microbiome of ruminants. Microbiome 2021, 9, 137. [Google Scholar] [CrossRef]
- Mani, S.; Aiyegoro, O.A.; Adeleke, M.A. Association between host genetics of sheep and the rumen microbial composition. Trop. Anim. Health Prod. 2022, 54, 109. [Google Scholar] [CrossRef]
- Lv, W.; Liu, X.; Sha, Y.; Shi, H.; Wei, H.; Luo, Y.; Wang, J.; Li, S.; Hu, J.; Guo, X.; et al. Rumen Fermentation-Microbiota-Host Gene Expression Interactions to Reveal the Adaptability of Tibetan Sheep in Different Periods. Animals 2021, 11, 3529. [Google Scholar] [CrossRef] [PubMed]
- Tanca, A.; Fraumene, C.; Manghina, V.; Palomba, A.; Abbondio, M.; Deligios, M.; Pagnozzi, D.; Addis, M.F.; Uzzau, S. Diversity and functions of the sheep faecal microbiota: A multi-omic characterization. Microb. Biotechnol. 2017, 10, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, K.; Zhang, C.; Feng, Y.; Zhang, X.; Wang, X.; Wu, G. Dynamics and stabilization of the rumen microbiome in yearling Tibetan sheep. Sci. Rep. 2019, 9, 19620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asanuma, N.; Yokoyama, S.; Hino, T. Effects of nitrate addition to a diet on fermentation and microbial populations in the rumen of goats, with special reference to Selenomonas ruminantium having the ability to reduce nitrate and nitrite. Anim. Sci. J. 2015, 86, 378–384. [Google Scholar] [CrossRef]
- Cremonesi, P.; Conte, G.; Severgnini, M.; Turri, F.; Monni, A.; Capra, E.; Rapetti, L.; Colombini, S.; Chessa, S.; Battelli, G.; et al. Evaluation of the effects of different diets on microbiome diversity and fatty acid composition of rumen liquor in dairy goat. Animal 2018, 12, 1856–1866. [Google Scholar] [CrossRef] [PubMed]
- Fliegerova, K.O.; Podmirseg, S.M.; Vinzelj, J.; Grilli, D.J.; Kvasnova, S.; Schierova, D.; Sechovcova, H.; Mrazek, J.; Siddi, G.; Arenas, G.N.; et al. The Effect of a High-Grain Diet on the Rumen Microbiome of Goats with a Special Focus on Anaerobic Fungi. Microorganisms 2021, 9, 157. [Google Scholar] [CrossRef]
- Peng, X.; Wilken, S.E.; Lankiewicz, T.S.; Gilmore, S.P.; Brown, J.L.; Henske, J.K.; Swift, C.L.; Salamov, A.; Barry, K.; Grigoriev, I.V.; et al. Genomic and functional analyses of fungal and bacterial consortia that enable lignocellulose breakdown in goat gut microbiomes. Nat. Microbiol. 2021, 6, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Min, B.R.; Solaiman, S.; Shange, R.; Eun, J.S. Gastrointestinal Bacterial and Methanogenic Archaea Diversity Dynamics Associated with Condensed Tannin-Containing Pine Bark Diet in Goats Using 16S rDNA Amplicon Pyrosequencing. Int. J. Microbiol. 2014, 2014, 141909. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhang, K.; Li, C.; Wang, X.; Chen, Y.; Yang, Y. Characterization and Comparison of Microbiota in the Gastrointestinal Tracts of the Goat (Capra hircus) During Preweaning Development. Front. Microbiol. 2019, 10, 2125. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Xu, Q.; Kong, F.; Yang, Y.; Wu, D.; Mishra, S.; Li, Y. Exploring the Goat Rumen Microbiome from Seven Days to Two Years. PLoS ONE 2016, 11, e0154354. [Google Scholar] [CrossRef]
- Zou, X.; Liu, G.; Meng, F.; Hong, L.; Li, Y.; Lian, Z.; Yang, Z.; Luo, C.; Liu, D. Exploring the Rumen and Cecum Microbial Community from Fetus to Adulthood in Goat. Animals 2020, 10, 1639. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, Y.; Yan, H.; Wang, X.; Qu, L.; Chen, Y. Rumen bacterial diversity of 80 to 110-day-old goats using 16S rRNA sequencing. PLoS ONE 2015, 10, e0117811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Cai, K.; Mishra, R.; Jha, R. In ovo supplementation of chitooligosaccharide and chlorella polysaccharide affects cecal microbial community, metabolic pathways, and fermentation metabolites in broiler chickens. Poult. Sci. 2020, 99, 4776–4785. [Google Scholar] [CrossRef] [PubMed]
- Palma-Hidalgo, J.M.; Jimenez, E.; Popova, M.; Morgavi, D.P.; Martin-Garcia, A.I.; Yanez-Ruiz, D.R.; Belanche, A. Inoculation with rumen fluid in early life accelerates the rumen microbial development and favours the weaning process in goats. Anim. Microbiome 2021, 3, 11. [Google Scholar] [CrossRef]
- Mammeri, M.; Obregon, D.A.; Chevillot, A.; Polack, B.; Julien, C.; Pollet, T.; Cabezas-Cruz, A.; Adjou, K.T. Cryptosporidium parvum Infection Depletes Butyrate Producer Bacteria in Goat Kid Microbiome. Front. Microbiol. 2020, 11, 548737. [Google Scholar] [CrossRef]
- Tong, J.; Ma, W.; Yang, R.; Wang, T.; Chen, X.; Zhang, X.; Tang, X.; Wen, Y.; Chang, J.; Chen, D. Dysbiosis of the gut microbiota maybe exacerbate orf pathology by promoting inflammatory immune responses. Vet. Microbiol. 2020, 251, 108884. [Google Scholar] [CrossRef]
- Wang, L.; Jin, L.; Xue, B.; Wang, Z.; Peng, Q. Characterizing the bacterial community across the gastrointestinal tract of goats: Composition and potential function. Microbiologyopen 2019, 8, e00820. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Huo, D.; You, Z.; Peng, Q.; Ma, C.; Chang, H.; Lin, X.; Wang, L.; Zhang, J. The distal intestinal microbiome of hybrids of Hainan black goats and Saanen goats. PLoS ONE 2020, 15, e0228496. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Shah, A.M.; Liu, Y.; Jin, L.; Wang, Z.; Xue, B.; Peng, Q. Relationship between true digestibility of dietary phosphorus and gastrointestinal bacteria of goats. PLoS ONE 2020, 15, e0225018. [Google Scholar] [CrossRef]
- Ren, Z.; Yao, R.; Liu, Q.; Deng, Y.; Shen, L.; Deng, H.; Zuo, Z.; Wang, Y.; Deng, J.; Cui, H.; et al. Effects of antibacterial peptides on rumen fermentation function and rumen microorganisms in goats. PLoS ONE 2019, 14, e0221815. [Google Scholar] [CrossRef]
- Zhuang, Y.; Chai, J.; Cui, K.; Bi, Y.; Diao, Q.; Huang, W.; Usdrowski, H.; Zhang, N. Longitudinal Investigation of the Gut Microbiota in Goat Kids from Birth to Postweaning. Microorganisms 2020, 8, 1111. [Google Scholar] [CrossRef] [PubMed]
- Dou, S.; Gadonna-Widehem, P.; Rome, V.; Hamoudi, D.; Rhazi, L.; Lakhal, L.; Larcher, T.; Bahi-Jaber, N.; Pinon-Quintana, A.; Guyonvarch, A.; et al. Characterisation of Early-Life Fecal Microbiota in Susceptible and Healthy Pigs to Post-Weaning Diarrhoea. PLoS ONE 2017, 12, e0169851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, L.; Estelle, J.; Kiilerich, P.; Ramayo-Caldas, Y.; Xia, Z.; Feng, Q.; Liang, S.; Pedersen, A.O.; Kjeldsen, N.J.; Liu, C.; et al. A reference gene catalogue of the pig gut microbiome. Nat. Microbiol. 2016, 1, 16161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Tsai, T.; Deng, F.; Wei, X.; Chai, J.; Knapp, J.; Apple, J.; Maxwell, C.V.; Lee, J.A.; Li, Y.; et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 2019, 7, 109. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Xiang, Y.; Wang, X.; Dai, B.; Zhang, X.; Ma, L.; Yang, H.; Lyu, W. Exploring the Possible Link between the Gut Microbiome and Fat Deposition in Pigs. Oxid. Med. Cell. Longev. 2022, 2022, 1098892. [Google Scholar] [CrossRef] [PubMed]
- Frese, S.A.; Parker, K.; Calvert, C.C.; Mills, D.A. Diet shapes the gut microbiome of pigs during nursing and weaning. Microbiome 2015, 3, 28. [Google Scholar] [CrossRef] [Green Version]
- Klinsoda, J.; Votterl, J.; Koger, S.; Metzler-Zebeli, B.U. Dietary Phytase- and Lactic Acid-Treated Cereals Caused Greater Taxonomic Adaptations than Functional Adaptations in the Cecal Metagenome of Growing Pigs. Appl. Environ. Microbiol. 2020, 87, e02240-20. [Google Scholar] [CrossRef]
- Petry, A.L.; Patience, J.F.; Koester, L.R.; Huntley, N.F.; Bedford, M.R.; Schmitz-Esser, S. Xylanase modulates the microbiota of ileal mucosa and digesta of pigs fed corn-based arabinoxylans likely through both a stimbiotic and prebiotic mechanism. PLoS ONE 2021, 16, e0246144. [Google Scholar] [CrossRef]
- Pollock, J.; Muwonge, A.; Hutchings, M.R.; Mainda, G.; Bronsvoort, B.M.; Gally, D.L.; Corbishley, A. Resistance to change: AMR gene dynamics on a commercial pig farm with high antimicrobial usage. Sci. Rep. 2020, 10, 1708. [Google Scholar] [CrossRef] [Green Version]
- Crespo-Piazuelo, D.; Estelle, J.; Revilla, M.; Criado-Mesas, L.; Ramayo-Caldas, Y.; Ovilo, C.; Fernandez, A.I.; Ballester, M.; Folch, J.M. Characterization of bacterial microbiota compositions along the intestinal tract in pigs and their interactions and functions. Sci. Rep. 2018, 8, 12727. [Google Scholar] [CrossRef]
- De Rodas, B.; Youmans, B.P.; Danzeisen, J.L.; Tran, H.; Johnson, T.J. Microbiome profiling of commercial pigs from farrow to finish. J. Anim. Sci. 2018, 96, 1778–1794. [Google Scholar] [CrossRef]
- Motta, V.; Trevisi, P.; Bertolini, F.; Ribani, A.; Schiavo, G.; Fontanesi, L.; Bosi, P. Exploring gastric bacterial community in young pigs. PLoS ONE 2017, 12, e0173029. [Google Scholar] [CrossRef]
- Verbeek, E.; Keeling, L.; Landberg, R.; Lindberg, J.E.; Dicksved, J. The gut microbiota and microbial metabolites are associated with tail biting in pigs. Sci. Rep. 2021, 11, 20547. [Google Scholar] [CrossRef]
- Borewicz, K.A.; Kim, H.B.; Singer, R.S.; Gebhart, C.J.; Sreevatsan, S.; Johnson, T.; Isaacson, R.E. Changes in the Porcine Intestinal Microbiome in Response to Infection with Salmonella enterica and Lawsonia intracellularis. PLoS ONE 2015, 10, e0139106. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Chen, J.; Gan, M.; Chen, L.; Zhao, Y.; Zhu, Y.; Niu, L.; Zhang, S.; Zhu, L.; Shen, L. Gut Microbiota Composition and Diversity in Different Commercial Swine Breeds in Early and Finishing Growth Stages. Animals 2022, 12, 1607. [Google Scholar] [CrossRef] [PubMed]
- Han, G.G.; Lee, J.Y.; Jin, G.D.; Park, J.; Choi, Y.H.; Chae, B.J.; Kim, E.B.; Choi, Y.J. Evaluating the association between body weight and the intestinal microbiota of weaned piglets via 16S rRNA sequencing. Appl. Microbiol. Biotechnol. 2017, 101, 5903–5911. [Google Scholar] [CrossRef] [PubMed]
- Cremonesi, P.; Biscarini, F.; Castiglioni, B.; Sgoifo, C.A.; Compiani, R.; Moroni, P. Gut microbiome modifications over time when removing in-feed antibiotics from the prophylaxis of post-weaning diarrhea in piglets. PLoS ONE 2022, 17, e0262199. [Google Scholar] [CrossRef] [PubMed]
- Holman, D.B.; Gzyl, K.E.; Mou, K.T.; Allen, H.K. Weaning Age and Its Effect on the Development of the Swine Gut Microbiome and Resistome. Msystems 2021, 6, e0068221. [Google Scholar] [CrossRef]
- Gaio, D.; DeMaere, M.Z.; Anantanawat, K.; Chapman, T.A.; Djordjevic, S.P.; Darling, A.E. Post-weaning shifts in microbiome composition and metabolism revealed by over 25 000 pig gut metagenome-assembled genomes. Microb. Genom. 2021, 7, 000501. [Google Scholar] [CrossRef]
- Guevarra, R.B.; Hong, S.H.; Cho, J.H.; Kim, B.R.; Shin, J.; Lee, J.H.; Kang, B.N.; Kim, Y.H.; Wattanaphansak, S.; Isaacson, R.E.; et al. The dynamics of the piglet gut microbiome during the weaning transition in association with health and nutrition. J Anim. Sci. Biotechnol. 2018, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Fenske, G.J.; Ghimire, S.; Antony, L.; Christopher-Hennings, J.; Scaria, J. Integration of culture-dependent and independent methods provides a more coherent picture of the pig gut microbiome. FEMS Microbiol. Ecol. 2020, 96, fiaa022. [Google Scholar] [CrossRef]
- Munk, P.; Andersen, V.D.; de Knegt, L.; Jensen, M.S.; Knudsen, B.E.; Lukjancenko, O.; Mordhorst, H.; Clasen, J.; Agerso, Y.; Folkesson, A.; et al. A sampling and metagenomic sequencing-based methodology for monitoring antimicrobial resistance in swine herds. J. Antimicrob. Chemother. 2017, 72, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gweon, H.S.; Shaw, L.P.; Swann, J.; De Maio, N.; AbuOun, M.; Niehus, R.; Hubbard, A.T.M.; Bowes, M.J.; Bailey, M.J.; Peto, T.E.A.; et al. The impact of sequencing depth on the inferred taxonomic composition and AMR gene content of metagenomic samples. Environ. Microbiome 2019, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wylezich, C.; Belka, A.; Hanke, D.; Beer, M.; Blome, S.; Hoper, D. Metagenomics for broad and improved parasite detection: A proof-of-concept study using swine faecal samples. Int. J. Parasitol. 2019, 49, 769–777. [Google Scholar] [CrossRef]
- Ramayo-Caldas, Y.; Prenafeta-Boldu, F.; Zingaretti, L.M.; Gonzalez-Rodriguez, O.; Dalmau, A.; Quintanilla, R.; Ballester, M. Gut eukaryotic communities in pigs: Diversity, composition and host genetics contribution. Anim. Microbiome 2020, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Mann, E.; Schmitz-Esser, S.; Zebeli, Q.; Wagner, M.; Ritzmann, M.; Metzler-Zebeli, B.U. Mucosa-associated bacterial microbiome of the gastrointestinal tract of weaned pigs and dynamics linked to dietary calcium-phosphorus. PLoS ONE 2014, 9, e86950. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Xin, Y.; Ma, Y.; Xu, X.; Zhao, S.; Cao, J. Screening of Microbes Associated With Swine Growth and Fat Deposition Traits Across the Intestinal Tract. Front. Microbiol. 2020, 11, 586776. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hu, H.; Zijlstra, R.T.; Zheng, J.; Gänzle, M.G. Metagenomic reconstructions of gut microbial metabolism in weanling pigs. Microbiome 2019, 7, 48. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wei, S.; Chen, N.; Xiang, Y.; Wang, Y.; Jin, M. Characteristics of gut microbiota in pigs with different breeds, growth periods and genders. Microb. Biotechnol. 2022, 15, 793–804. [Google Scholar] [CrossRef]
- Wylensek, D.; Hitch, T.; Riedel, T.; Afrizal, A.; Kumar, N.; Wortmann, E.; Liu, T.; Devendran, S.; Lesker, T.R.; Hernández, S.B.; et al. A collection of bacterial isolates from the pig intestine reveals functional and taxonomic diversity. Nat. Commun. 2020, 11, 6389. [Google Scholar] [CrossRef]
- Qu, A.; Brulc, J.M.; Wilson, M.K.; Law, B.F.; Theoret, J.R.; Joens, L.A.; Konkel, M.E.; Angly, F.; Dinsdale, E.A.; Edwards, R.A.; et al. Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS ONE 2008, 3, e2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danzeisen, J.L.; Kim, H.B.; Isaacson, R.E.; Tu, Z.J.; Johnson, T.J. Modulations of the chicken cecal microbiome and metagenome in response to anticoccidial and growth promoter treatment. PLoS ONE 2011, 6, e27949. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; Lee, T.K.; Sul, W.J. Metagenomic Analysis of Chicken Gut Microbiota for Improving Metabolism and Health of Chickens—A Review. Asian-Australas. J. Anim. Sci. 2015, 28, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- Durazzi, F.; Sala, C.; Castellani, G.; Manfreda, G.; Remondini, D.; De Cesare, A. Comparison between 16S rRNA and shotgun sequencing data for the taxonomic characterization of the gut microbiota. Sci. Rep. 2021, 11, 3030. [Google Scholar] [CrossRef]
- De Cesare, A.; Sirri, F.; Manfreda, G.; Moniaci, P.; Giardini, A.; Zampiga, M.; Meluzzi, A. Effect of dietary supplementation with Lactobacillus acidophilus D2/CSL (CECT 4529) on caecum microbioma and productive performance in broiler chickens. PLoS ONE 2017, 12, e0176309. [Google Scholar] [CrossRef] [Green Version]
- Gilroy, R.; Ravi, A.; Getino, M.; Pursley, I.; Horton, D.L.; Alikhan, N.F.; Baker, D.; Gharbi, K.; Hall, N.; Watson, M.; et al. Extensive microbial diversity within the chicken gut microbiome revealed by metagenomics and culture. PeerJ 2021, 9, e10941. [Google Scholar] [CrossRef] [PubMed]
- Medvecky, M.; Cejkova, D.; Polansky, O.; Karasova, D.; Kubasova, T.; Cizek, A.; Rychlik, I. Whole genome sequencing and function prediction of 133 gut anaerobes isolated from chicken caecum in pure cultures. BMC Genom. 2018, 19, 561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Shi, Y.; Liu, P.; Yang, Y.; Zhou, C.; Li, G.; Luo, J.; Zhang, C.; Cao, H.; Hu, G.; et al. 16S rRNA gene sequencing reveals an altered composition of the gut microbiota in chickens infected with a nephropathogenic infectious bronchitis virus. Sci. Rep. 2020, 10, 3556. [Google Scholar] [CrossRef] [Green Version]
- Clavijo, V.; Florez, M.J.V. The gastrointestinal microbiome and its association with the control of pathogens in broiler chicken production: A review. Poult. Sci. 2018, 97, 1006–1021. [Google Scholar] [CrossRef]
- Such, N.; Farkas, V.; Csitari, G.; Pal, L.; Marton, A.; Menyhart, L.; Dublecz, K. Relative Effects of Dietary Administration of a Competitive Exclusion Culture and a Synbiotic Product, Age and Sampling Site on Intestinal Microbiota Maturation in Broiler Chickens. Vet. Sci. 2021, 8, 187. [Google Scholar] [CrossRef]
- Adenaike, A.S.; Akpan, U.; Awopejo, O.O.; Oloye, O.S.; Alli-Balogun, A.O.; Agbaje, M.; Ikeobi, C.O.N. Characterization of the cecal microbiome composition of Nigerian indigenous chickens. Trop. Anim. Health Prod. 2022, 54, 211. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Cho, S.; La, T.M.; Lee, H.J.; Lee, J.B.; Park, S.Y.; Song, C.S.; Choi, I.S.; Lee, S.W. Comparison of microbiota in the cloaca, colon, and magnum of layer chicken. PLoS ONE 2020, 15, e0237108. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xiang, H.; Zhang, H.; Zhu, X.; Wang, D.; Wang, J.; Yin, T.; Liu, L.; Kong, M.; Li, H.; et al. Rearing system causes changes of behavior, microbiome, and gene expression of chickens. Poult. Sci. 2019, 98, 3365–3376. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liao, X.P.; D’Souza, A.W.; Boolchandani, M.; Li, S.H.; Cheng, K.; Luis Martinez, J.; Li, L.; Feng, Y.J.; Fang, L.X.; et al. Environmental remodeling of human gut microbiota and antibiotic resistome in livestock farms. Nat. Commun. 2020, 11, 1427. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, G.A.; Richardson, E.; Clark, J.; Keshri, J.; Drechsler, Y.; Berrang, M.E.; Meinersmann, R.J.; Cox, N.A.; Oakley, B.B. Broiler chickens and early life programming: Microbiome transplant-induced cecal community dynamics and phenotypic effects. PLoS ONE 2020, 15, e0242108. [Google Scholar] [CrossRef]
- Kabir, M.H.B.; Han, Y.; Lee, S.H.; Nugraha, A.B.; Recuenco, F.; Murakoshi, F.; Xuan, X.; Kato, K. Prevalence and molecular characterization of Cryptosporidium species in poultry in Bangladesh. One Health 2020, 9, 100122. [Google Scholar] [CrossRef]
- Wei, S.; Morrison, M.; Yu, Z. Bacterial census of poultry intestinal microbiome. Poult. Sci. 2013, 92, 671–683. [Google Scholar] [CrossRef]
- Saxena, S.; Saxena, V.; Tomar, S.; Sapcota, D.; Gonmei, G. Characterisation of caecum and crop microbiota of Indian indigenous chicken targeting multiple hypervariable regions within 16S rRNA gene. Br. Poult. Sci. 2016, 57, 381–389. [Google Scholar] [CrossRef]
- Biasato, I.; Ferrocino, I.; Grego, E.; Dabbou, S.; Gai, F.; Gasco, L.; Cocolin, L.; Capucchio, M.T.; Schiavone, A. Gut Microbiota and Mucin Composition in Female Broiler Chickens Fed Diets including Yellow Mealworm (Tenebrio molitor, L.). Animals 2019, 9, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Diarra, M.S.; Choi, J.; Rodas-Gonzalez, A.; Lepp, D.; Liu, S.; Lu, P.; Mogire, M.; Gong, J.; Wang, Q.; et al. Effects of encapsulated cinnamaldehyde on growth performance, intestinal digestive and absorptive functions, meat quality and gut microbiota in broiler chickens. Transl. Anim. Sci. 2021, 5, txab099. [Google Scholar] [CrossRef]
- Videnska, P.; Faldynova, M.; Juricova, H.; Babak, V.; Sisak, F.; Havlickova, H.; Rychlik, I. Chicken faecal microbiota and disturbances induced by single or repeated therapy with tetracycline and streptomycin. BMC Vet. Res. 2013, 9, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Costa, V.M.; McGrann, K.M.; Hughes, D.W.; Wright, G.D. Sampling the antibiotic resistome. Science 2006, 311, 374–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skandalis, N.; Maeusli, M.; Papafotis, D.; Miller, S.; Lee, B.; Theologidis, I.; Luna, B. Environmental Spread of Antibiotic Resistance. Antibiotics 2021, 10, 640. [Google Scholar] [CrossRef]
- Kim, D.W.; Cha, C.J. Antibiotic resistome from the One-Health perspective: Understanding and controlling antimicrobial resistance transmission. Exp. Mol. Med. 2021, 53, 301–309. [Google Scholar] [CrossRef]
- Liu, J.; Taft, D.H.; Maldonado-Gomez, M.X.; Johnson, D.; Treiber, M.L.; Lemay, D.G.; DePeters, E.J.; Mills, D.A. The fecal resistome of dairy cattle is associated with diet during nursing. Nat. Commun. 2019, 10, 4406. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Catalina, A.; Atxaerandio, R.; Garcia-Rodriguez, A.; Goiri, I.; Gutierrez-Rivas, M.; Jimenez-Montero, J.A.; Gonzalez-Recio, O. Characterisation of the rumen resistome in Spanish dairy cattle. Anim. Microbiome 2021, 3, 63. [Google Scholar] [CrossRef]
- Ma, T.; McAllister, T.A.; Guan, L.L. A review of the resistome within the digestive tract of livestock. J. Anim. Sci. Biotechnol. 2021, 12, 121. [Google Scholar] [CrossRef]
- Hitch, T.C.A.; Thomas, B.J.; Friedersdorff, J.C.A.; Ougham, H.; Creevey, C.J. Deep sequence analysis reveals the ovine rumen as a reservoir of antibiotic resistance genes. Environ. Pollut. 2018, 235, 571–575. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, Y.; Liu, F.; Cao, J.; Lv, N.; Zhu, B.; Zhang, G.; Gao, G.F. Integrated metagenomic and metatranscriptomic profiling reveals differentially expressed resistomes in human, chicken, and pig gut microbiomes. Environ. Int. 2020, 138, 105649. [Google Scholar] [CrossRef]
- Koorakula, R.; Schiavinato, M.; Ghanbari, M.; Wegl, G.; Grabner, N.; Koestelbauer, A.; Klose, V.; Dohm, J.C.; Domig, K.J. Metatranscriptomic Analysis of the Chicken Gut Resistome Response to In-Feed Antibiotics and Natural Feed Additives. Front. Microbiol. 2022, 13, 833790. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.; Salvatierra, G.; Davila-Barclay, A.; Ayzanoa, B.; Castillo-Vilcahuaman, C.; Huang, M.; Pajuelo, M.J.; Lescano, A.G.; Cabrera, L.; Calderon, M.; et al. Market Chickens as a Source of Antibiotic-Resistant Escherichia coli in a Peri-Urban Community in Lima, Peru. Front. Microbiol. 2021, 12, 635871. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotech. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2: An improved and extensible approach for metagenome inference. BioRxiv 2019, 672295. [Google Scholar] [CrossRef] [Green Version]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef]
- Wemheuer, F.; Taylor, J.A.; Daniel, R.; Johnston, E.; Meinicke, P.; Thomas, T.; Wemheuer, B. Tax4Fun2: A R-based tool for the rapid prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene marker gene sequences. Environ. Microbiome 2020, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Jones, R.B.; Fodor, A.A. Inference-based accuracy of metagenome prediction tools varies across sample types and functional categories. Microbiome 2020, 8, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toole, D.R.; Zhao, J.; Martens-Habbena, W.; Strauss, S.L. Bacterial functional prediction tools detect but underestimate metabolic diversity compared to shotgun metagenomics in southwest Florida soils. Appl. Soil Ecol. 2021, 168, 104129. [Google Scholar] [CrossRef]
- Sun, B.; Hou, L.; Yang, Y. The Development of the Gut Microbiota and Short-Chain Fatty Acids of Layer Chickens in Different Growth Periods. Front. Vet. Sci. 2021, 8, 666535. [Google Scholar] [CrossRef]
- Kogut, M.H.; Arsenault, R.J. Editorial: Gut Health: The New Paradigm in Food Animal Production. Front. Vet. Sci. 2016, 3, 71. [Google Scholar] [CrossRef] [Green Version]
- Kraimi, N.; Dawkins, M.; Gebhardt-Henrich, S.G.; Velge, P.; Rychlik, I.; Volf, J.; Creach, P.; Smith, A.; Colles, F.; Leterrier, C. Influence of the microbiota-gut-brain axis on behavior and welfare in farm animals: A review. Physiol. Behav. 2019, 210, 112658. [Google Scholar] [CrossRef]
- O’Hara, E.; Neves, A.; Song, Y.; Guan, L.L. The Role of the Gut Microbiome in Cattle Production and Health: Driver or Passenger? Annu. Rev. Anim. Biosci. 2020, 8, 199–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, J.; Reese, A.T. Possibilities and limits for using the gut microbiome to improve captive animal health. Anim. Microbiome 2021, 3, 89. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Luo, S.; Yan, C. Gut Microbiota Implications for Health and Welfare in Farm Animals: A Review. Animals 2022, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Li, D.; Leng, D.; Kui, H.; Bai, X.; Wang, T. Gut microbiota and meat quality. Front. Microbiol. 2022, 13, 951726. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, J.M.; Welch, C.B. Using microbiome information to understand and improve animal performance. Ital. J. Anim. Sci. 2022, 21, 899–913. [Google Scholar] [CrossRef]
- Khalil, A.; Batool, A.; Arif, S. Healthy Cattle Microbiome and Dysbiosis in Diseased Phenotypes. Ruminants 2022, 2, 134–156. [Google Scholar] [CrossRef]
- Clemmons, B.A.; Voy, B.H.; Myer, P.R. Altering the Gut Microbiome of Cattle: Considerations of Host-Microbiome Interactions for Persistent Microbiome Manipulation. Microb. Ecol. 2019, 77, 523–536. [Google Scholar] [CrossRef]
- Malmuthuge, N.; Guan, L.L. Understanding the gut microbiome of dairy calves: Opportunities to improve early-life gut health. J. Dairy Sci. 2017, 100, 5996–6005. [Google Scholar] [CrossRef] [Green Version]
- Campanaro, S.; Treu, L.; Kougias, P.G.; Zhu, X.; Angelidaki, I. Taxonomy of anaerobic digestion microbiome reveals biases associated with the applied high throughput sequencing strategies. Sci. Rep. 2018, 8, 1926. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forcina, G.; Pérez-Pardal, L.; Carvalheira, J.; Beja-Pereira, A. Gut Microbiome Studies in Livestock: Achievements, Challenges, and Perspectives. Animals 2022, 12, 3375. https://doi.org/10.3390/ani12233375
Forcina G, Pérez-Pardal L, Carvalheira J, Beja-Pereira A. Gut Microbiome Studies in Livestock: Achievements, Challenges, and Perspectives. Animals. 2022; 12(23):3375. https://doi.org/10.3390/ani12233375
Chicago/Turabian StyleForcina, Giovanni, Lucía Pérez-Pardal, Júlio Carvalheira, and Albano Beja-Pereira. 2022. "Gut Microbiome Studies in Livestock: Achievements, Challenges, and Perspectives" Animals 12, no. 23: 3375. https://doi.org/10.3390/ani12233375
APA StyleForcina, G., Pérez-Pardal, L., Carvalheira, J., & Beja-Pereira, A. (2022). Gut Microbiome Studies in Livestock: Achievements, Challenges, and Perspectives. Animals, 12(23), 3375. https://doi.org/10.3390/ani12233375