
Trans-Species Fecal Transplant Revealed the Role of the Gut Microbiome as a Contributor to Energy Metabolism and Development of Skeletal Muscle



Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Treatments
2.2. Preparation and Transplantation of Fecal Microbiota
2.3. Sample Collection
2.4. 16S rRNA Sequencing and Analysis
2.5. Histological Assessment of the Skeletal Muscle Fiber Morphology
2.6. Skeletal Muscle Gene Expression Analysis by RT–PCR
2.7. Glycogen Measurements in Skeletal Muscle
2.8. Statistical Analysis
3. Results
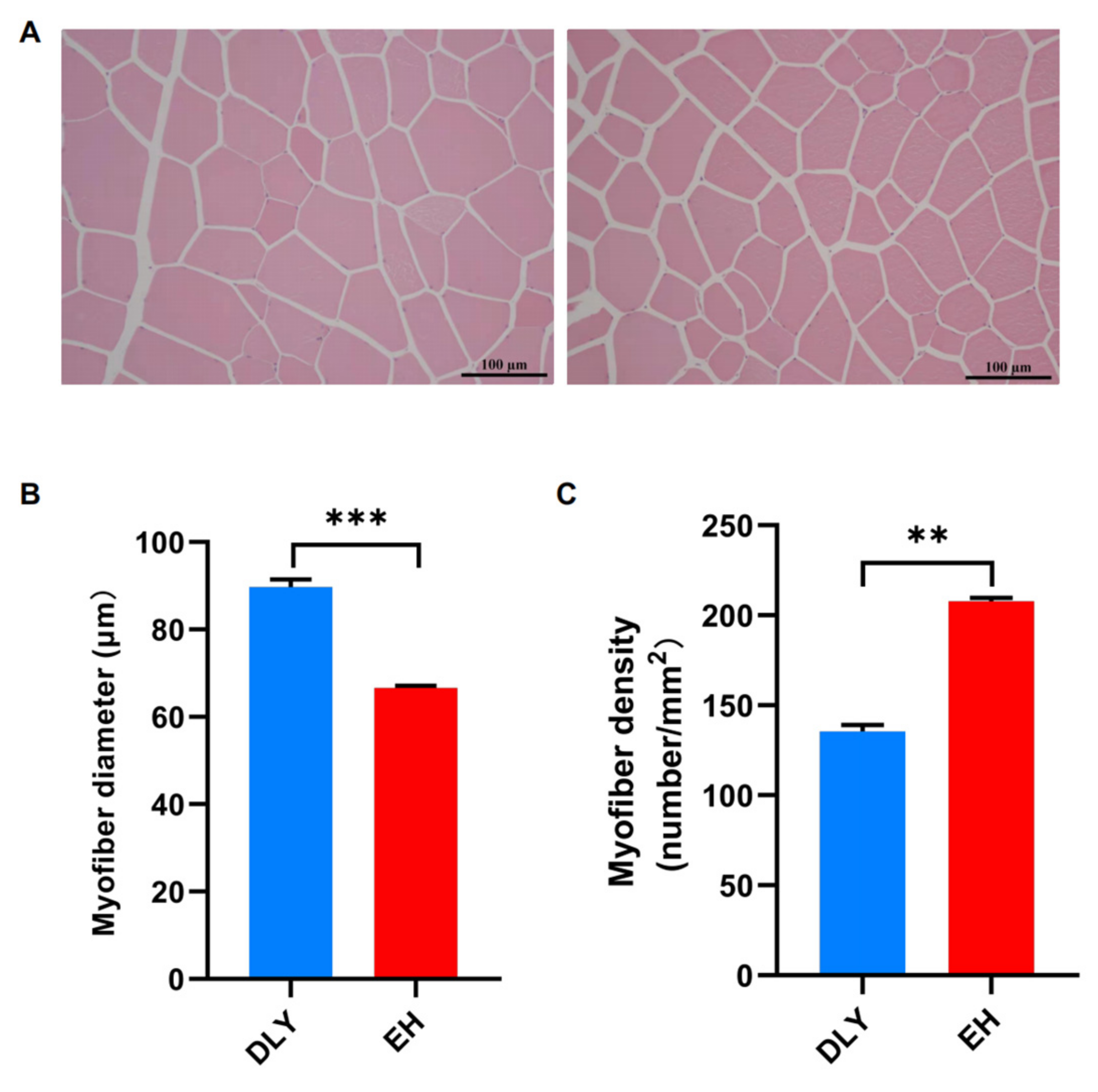
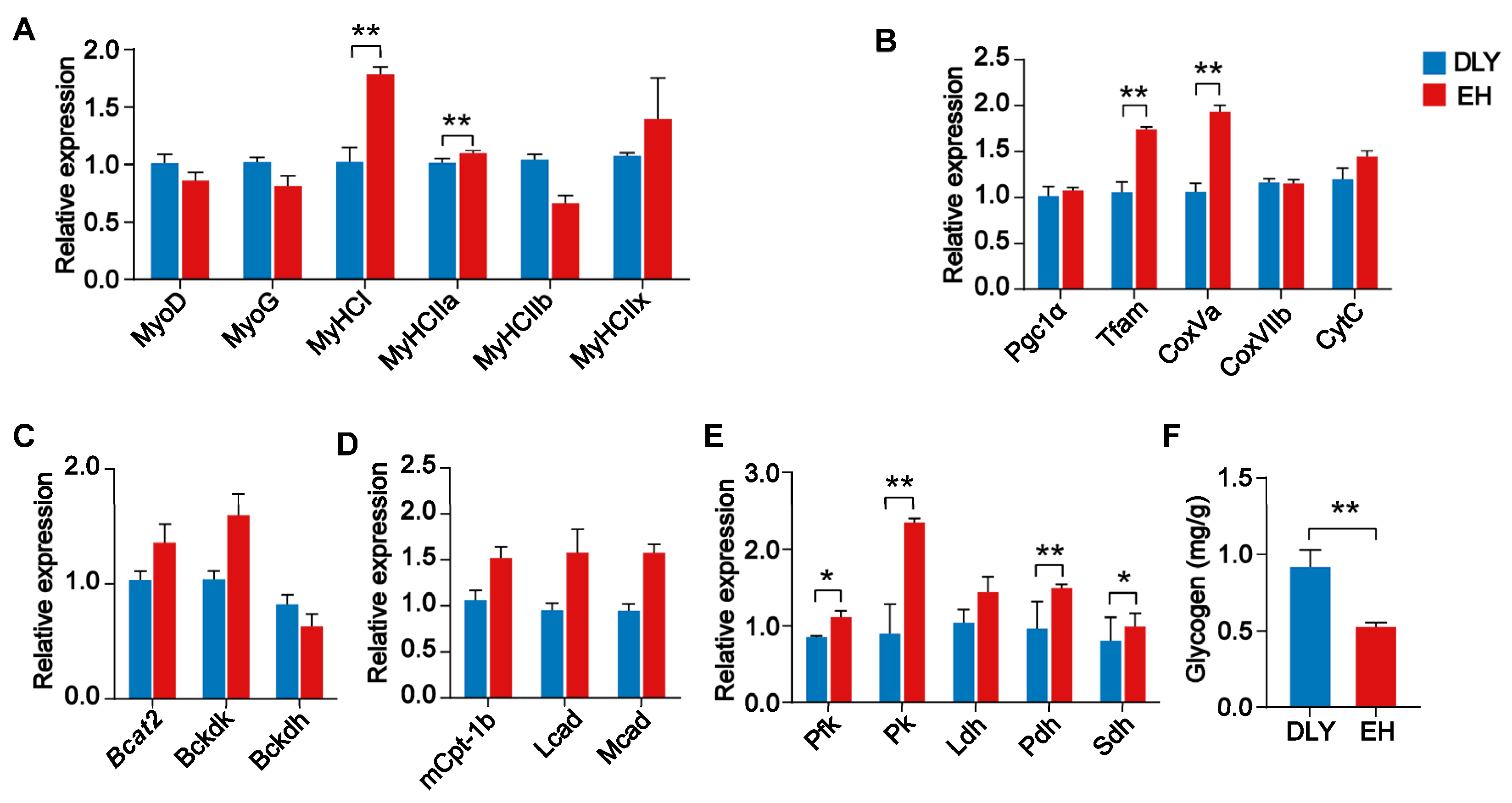
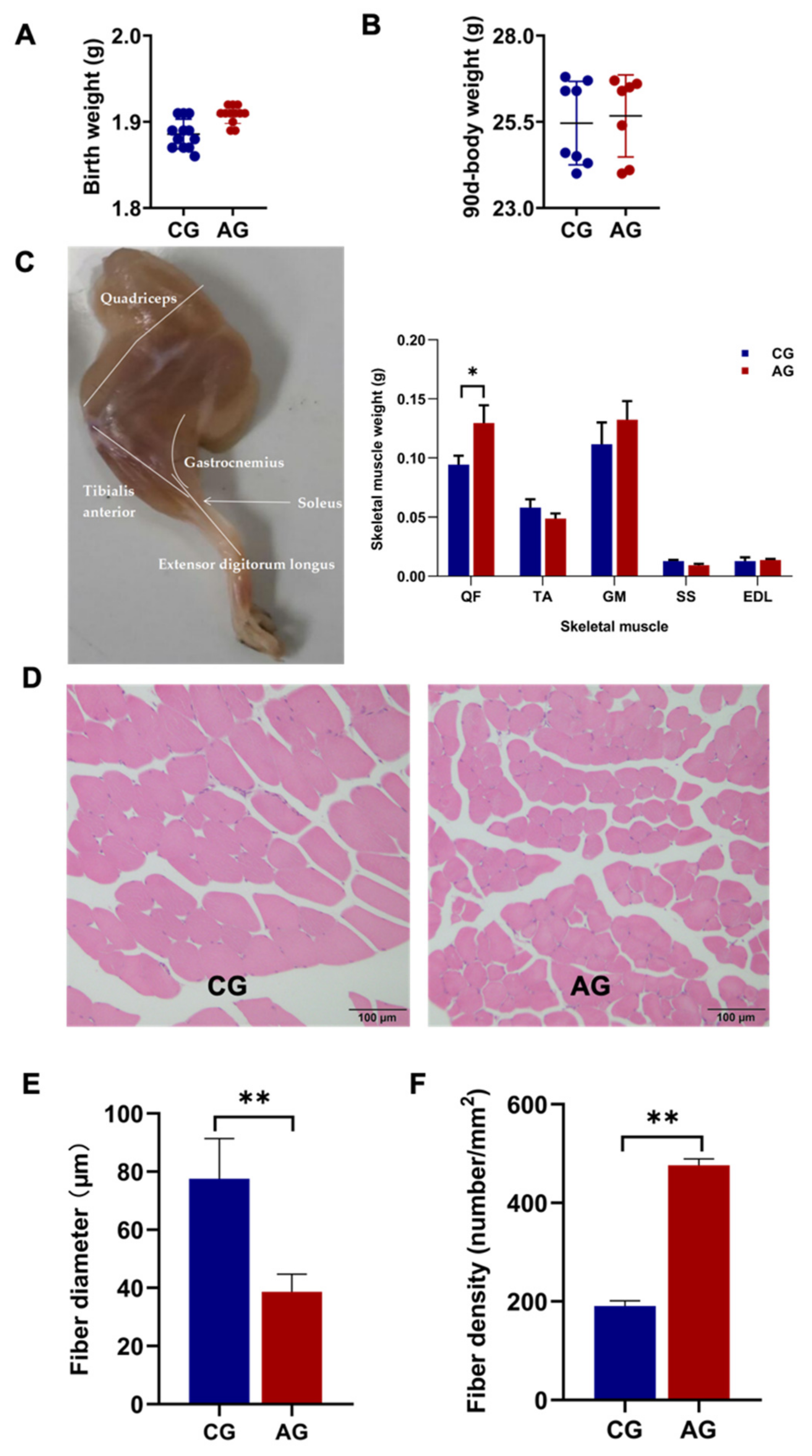
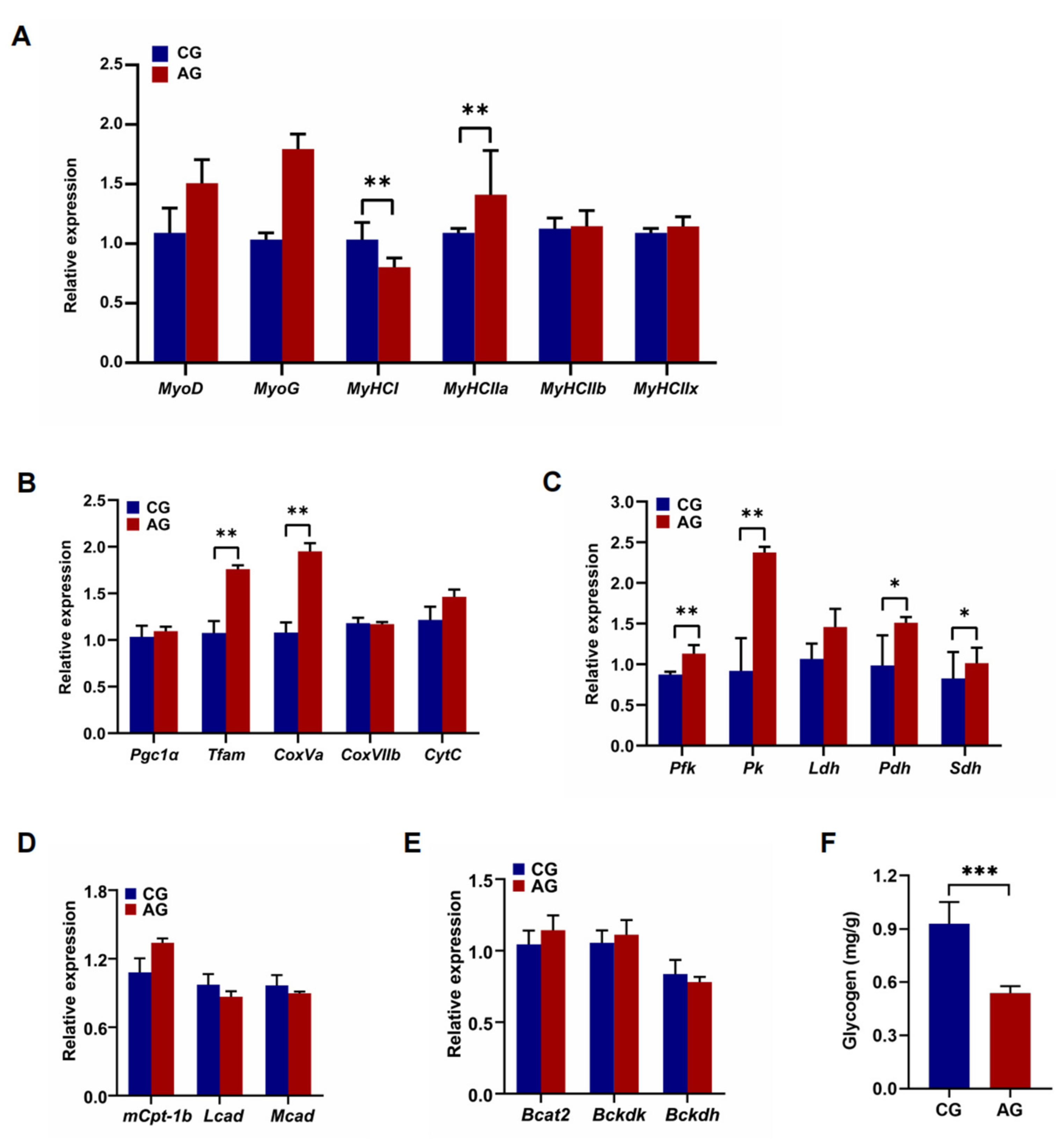
3.1. Fiber Size and Energy Metabolism Characteristics in the Skeletal Muscle of EH Pigs
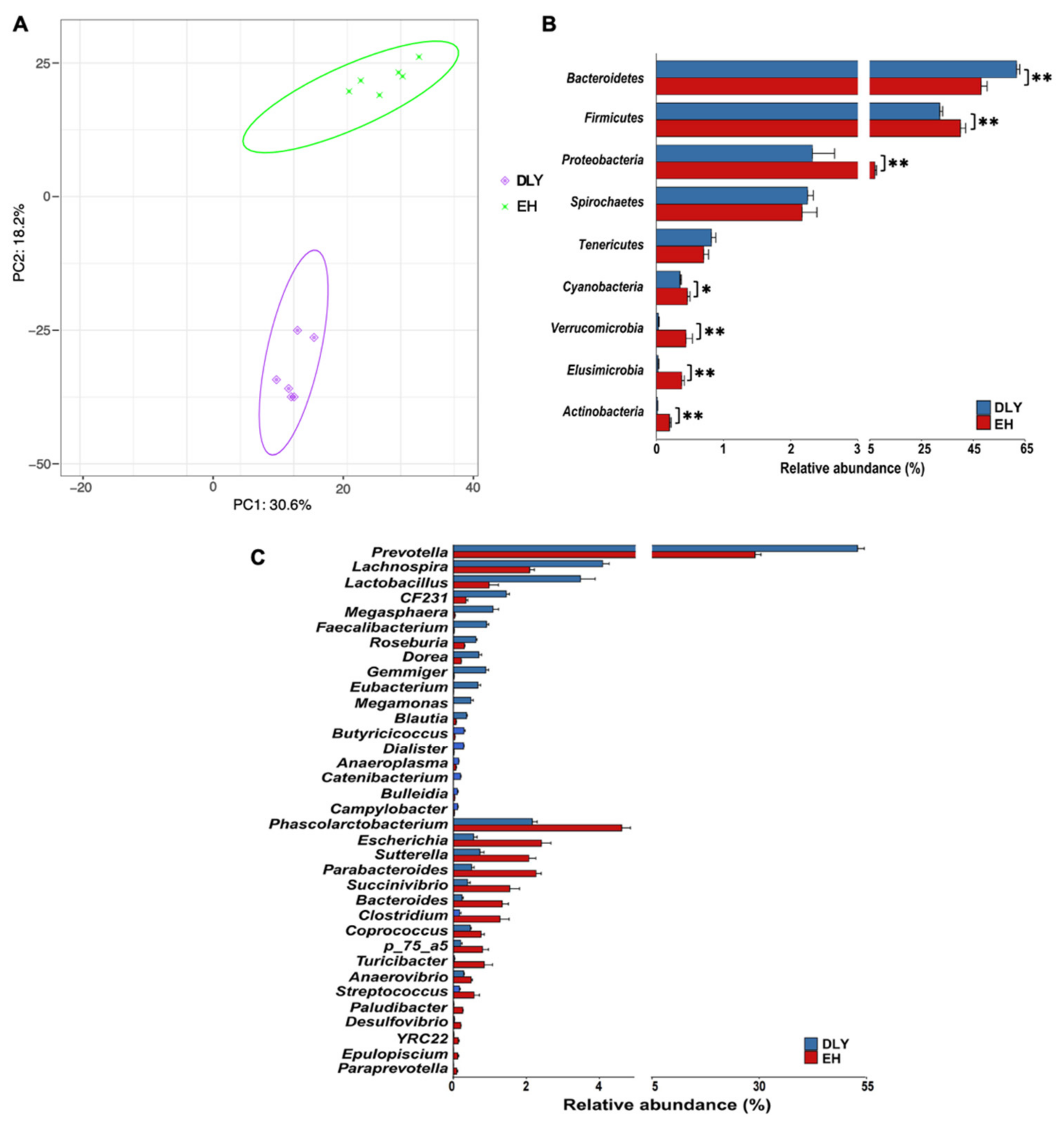
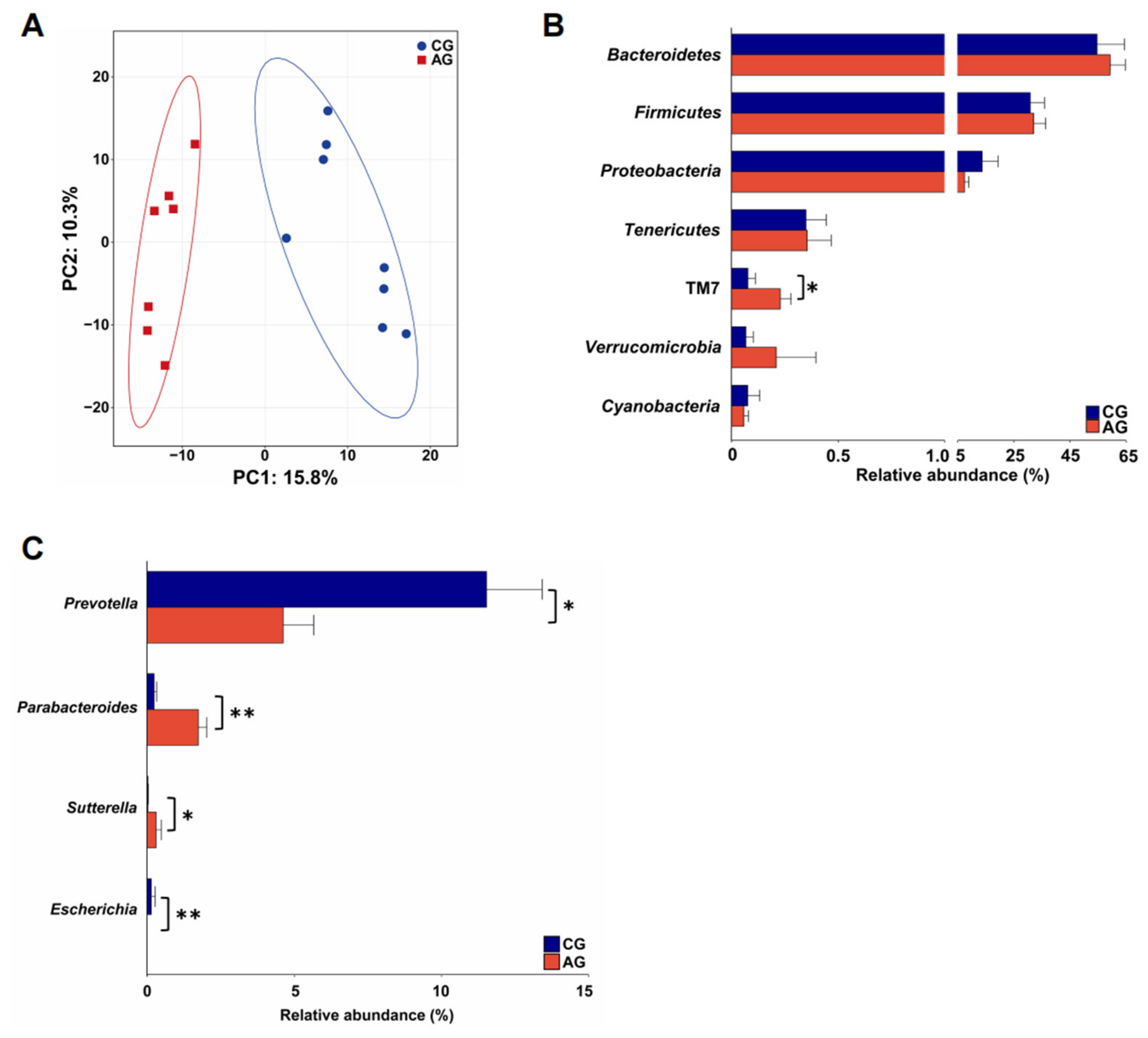
3.2. Composition and Function of the Gut Microbiota of EH Pigs
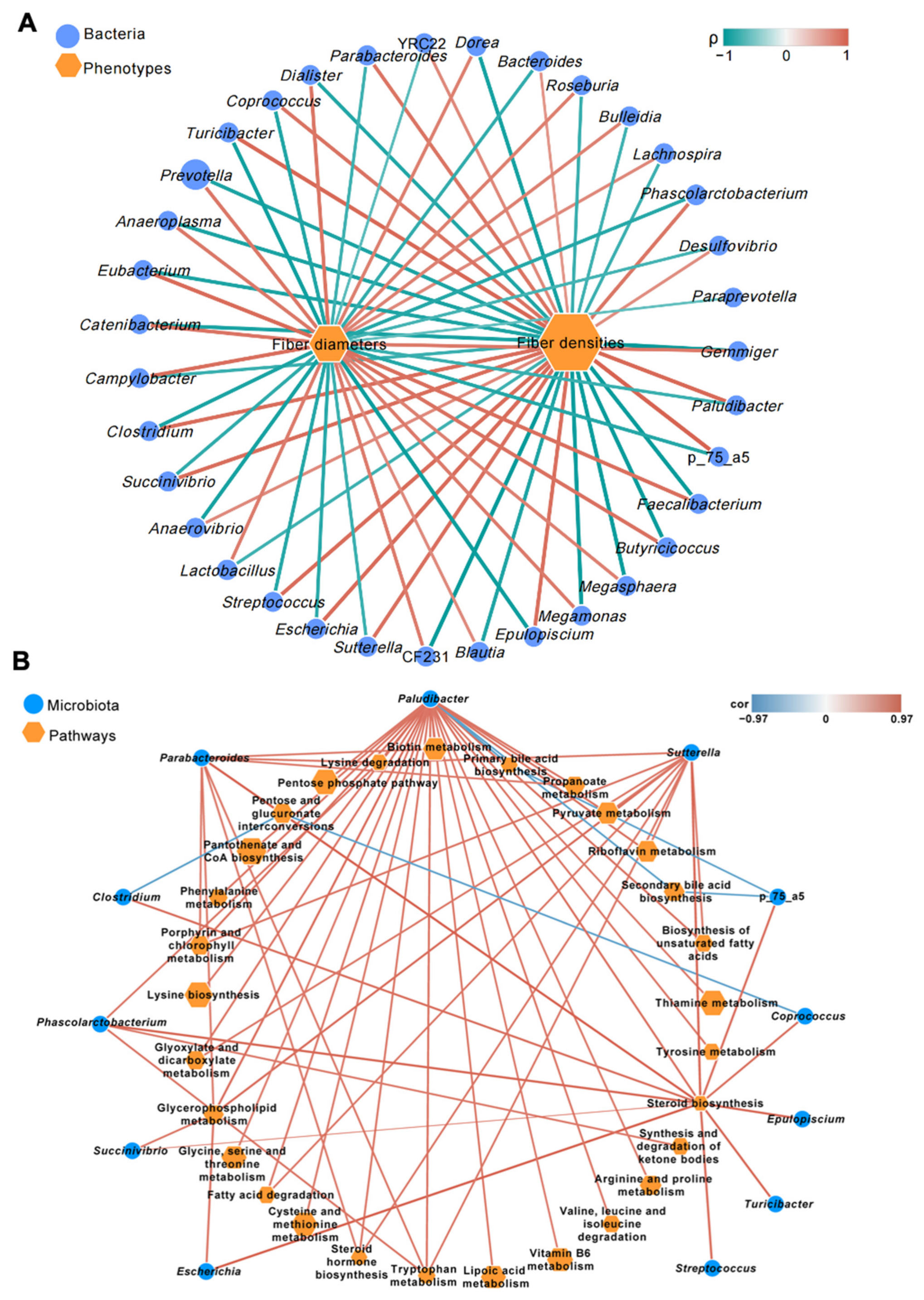
3.3. Correlations between Different Microbial Genera and Skeletal Muscle Phenotypes of Pigs
3.4. Gut Microbiota Transplantation Altered Fiber Size and Energy Metabolism in Mice Skeletal Muscle
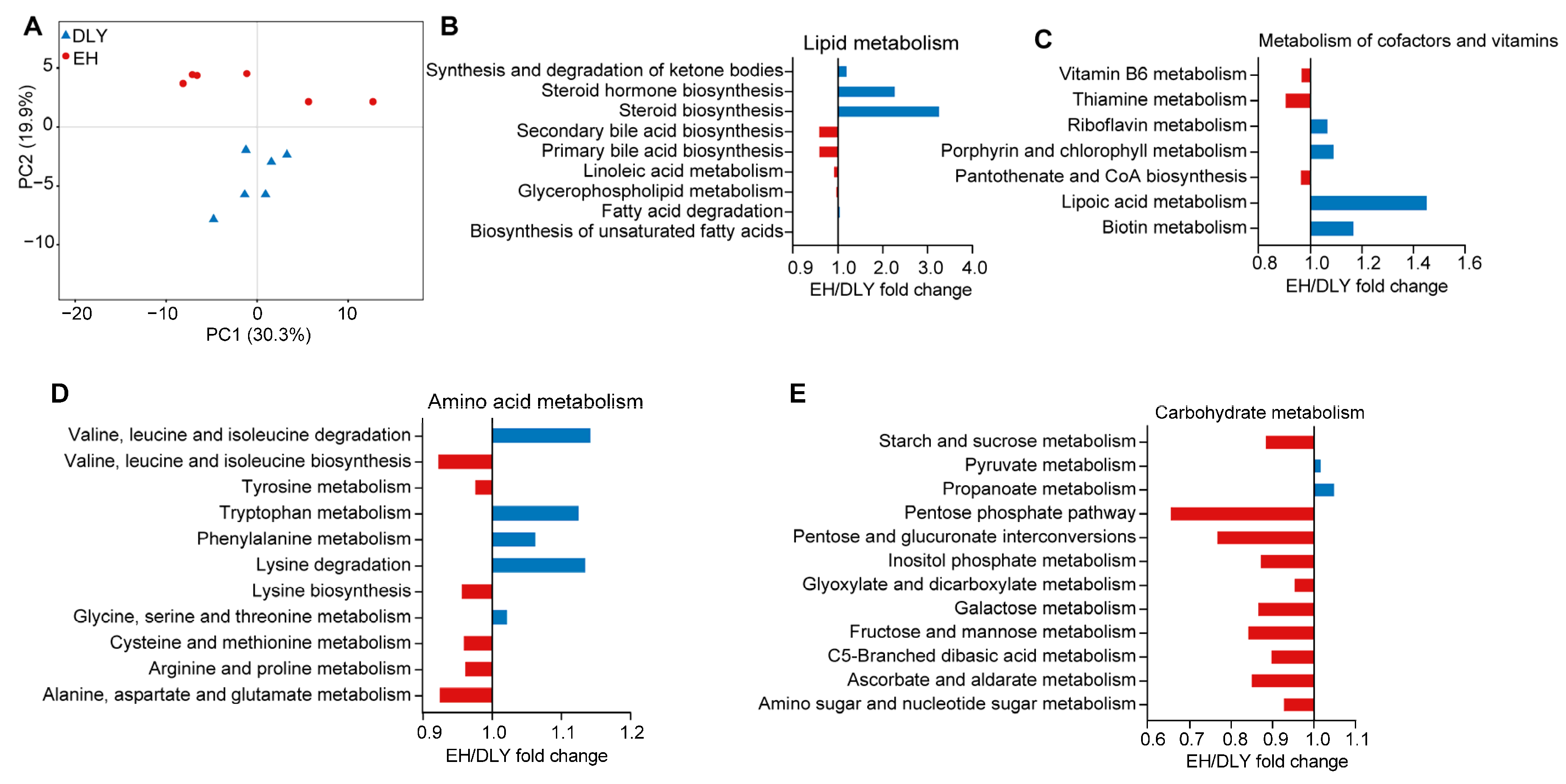
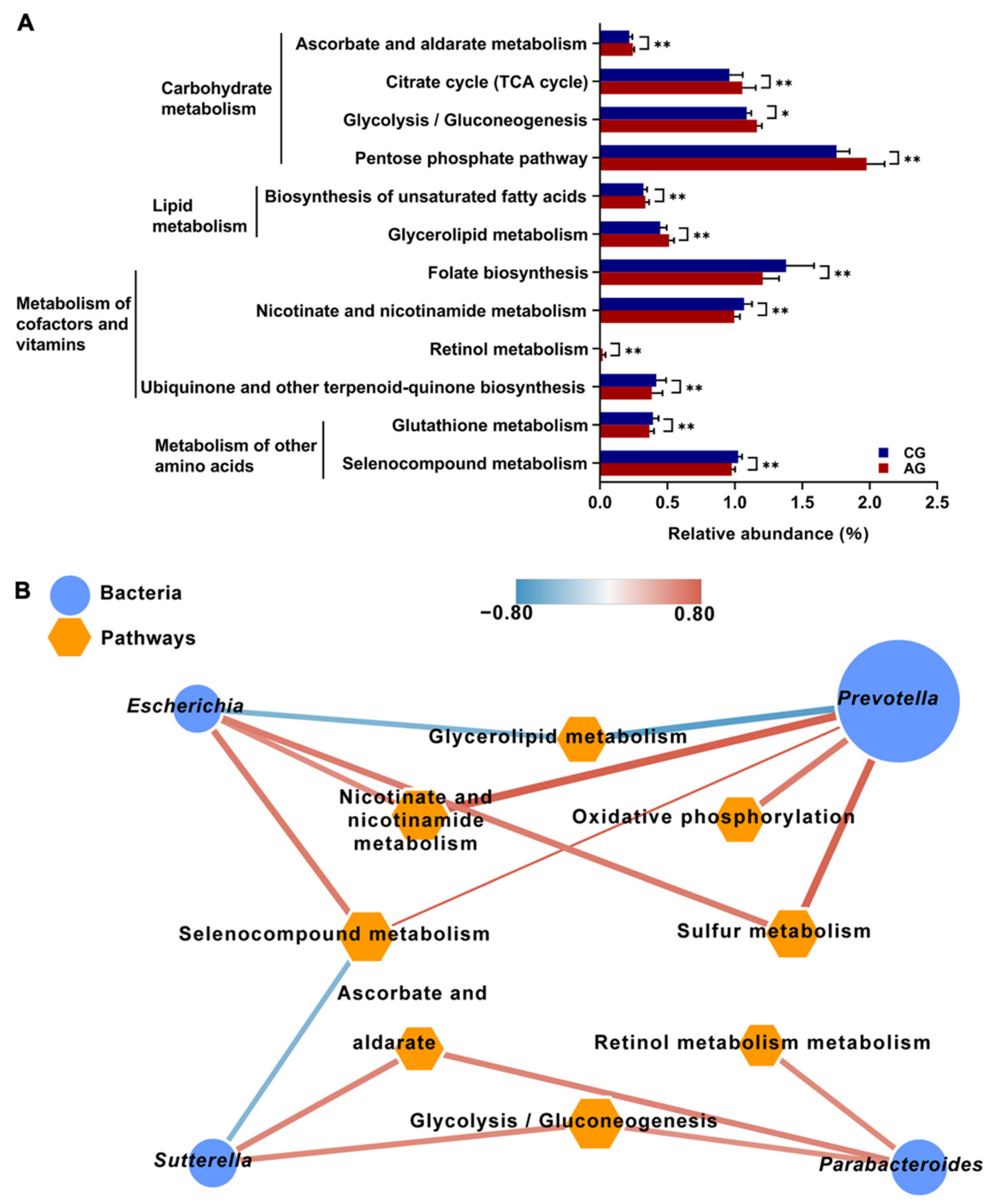
3.5. Gut Microbiota Composition and Functional Profiles of Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The Impact of the Gut Microbiota on Human Health: An Integrative View. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beller, L.; Deboutte, W.; Falony, G.; Vieira-Silva, S.; Tito, R.Y.; Valles-Colomer, M.; Rymenans, L.; Jansen, D.; Van Espen, L.; Papadaki, M.I.; et al. Successional Stages in Infant Gut Microbiota Maturation. mBio 2021, 12, e0185721. [Google Scholar] [CrossRef] [PubMed]
- Spor, A.; Koren, O.; Ley, R. Unravelling the Effects of the Environment and Host Genotype on the Gut Microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Schubert, A.M.; Zackular, J.P.; Iverson, K.D.; Young, V.B.; Petrosino, J.F. Stabilization of the Murine Gut Microbiome Following Weaning. Gut Microbes 2012, 3, 383–393. [Google Scholar] [CrossRef]
- Diao, H.; Yan, H.L.; Xiao, Y.; Yu, B.; Yu, J.; He, J.; Zheng, P.; Zeng, B.H.; Wei, H.; Mao, X.B.; et al. Intestinal Microbiota Could Transfer Host Gut Characteristics from Pigs to Mice. BMC Microbiol. 2016, 16, 238. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Diao, H.; Xiao, Y.; Li, W.; Yu, B.; He, J.; Yu, J.; Zheng, P.; Mao, X.; Luo, Y.; et al. Gut Microbiota Can Transfer Fiber Characteristics and Lipid Metabolic Profiles of Skeletal Muscle from Pigs to Germ-Free Mice. Sci. Rep. 2016, 6, 31786. [Google Scholar] [CrossRef] [Green Version]
- Lahiri, S.; Kim, H.; Garcia-Perez, I.; Reza, M.M.; Martin, K.A.; Kundu, P.; Cox, L.M.; Selkrig, J.; Posma, J.M.; Zhang, H.; et al. The Gut Microbiota Influences Skeletal Muscle Mass and Function in Mice. Sci. Transl. Med. 2019, 11, eaan5662. [Google Scholar] [CrossRef] [Green Version]
- Ticinesi, A.; Lauretani, F.; Milani, C.; Nouvenne, A.; Tana, C.; Del Rio, D.; Maggio, M.; Ventura, M.; Meschi, T. Aging Gut Microbiota at the Cross-Road between Nutrition, Physical Frailty, and Sarcopenia: Is There a Gut-Muscle Axis? Nutrients 2017, 9, 1303. [Google Scholar] [CrossRef] [Green Version]
- Han, P.; Li, P.; Zhou, W.; Fan, L.; Wang, B.; Liu, H.; Gao, C.; Du, T.; Pu, G.; Wu, C.; et al. Effects of Various Levels of Dietary Fiber on Carcass Traits, Meat Quality and Myosin Heavy Chain I, IIa, IIx and IIb Expression in Muscles in Erhualian and Large White Pigs. Meat Sci. 2020, 169, 108160. [Google Scholar] [CrossRef]
- Hu, J.; Chen, L.; Tang, Y.; Xie, C.; Xu, B.; Shi, M.; Zheng, W.; Zhou, S.; Wang, X.; Liu, L.; et al. Standardized Preparation for Fecal Microbiota Transplantation in Pigs. Front. Microbiol. 2018, 9, 1328. [Google Scholar] [CrossRef]
- Hu, J.; Ma, L.; Nie, Y.; Chen, J.; Zheng, W.; Wang, X.; Xie, C.; Zheng, Z.; Wang, Z.; Yang, T.; et al. A Microbiota-Derived Bacteriocin Targets the Host to Confer Diarrhea Resistance in Early-Weaned Piglets. Cell Host Microbe 2018, 24, 817–832.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME Improves Sensitivity and Speed of Chimera Detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and Clustering Orders of Magnitude Faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, T.J.; Huws, S.A.; Edwards, J.E.; Kingston-Smith, A.H.; Siu-Ting, K.; Hughes, M.; Rubino, F.; Friedersdorff, M.; Creevey, C.J. CowPI: A Rumen Microbiome Focussed Version of the PICRUSt Functional Inference Software. Front. Microbiol. 2018, 9, 1095. [Google Scholar] [CrossRef]
- Wiltafsky, M.K.; Pfaffl, M.W.; Roth, F.X. The Effects of Branched-Chain Amino Acid Interactions on Growth Performance, Blood Metabolites, Enzyme Kinetics and Transcriptomics in Weaned Pigs. Br. J. Nutr. 2010, 103, 964–976. [Google Scholar] [CrossRef] [Green Version]
- Ying, F.; Zhang, L.; Bu, G.; Xiong, Y.; Zuo, B. Muscle Fiber-Type Conversion in the Transgenic Pigs with Overexpression of PGC1α Gene in Muscle. Biochem. Biophys. Res. Commun. 2016, 480, 669–674. [Google Scholar] [CrossRef]
- Ramakers, C.; Ruijter, J.M.; Deprez, R.H.L.; Moorman, A.F.M. Assumption-Free Analysis of Quantitative Real-Time Polymerase Chain Reaction (PCR) Data. Neurosci. Lett. 2003, 339, 62–66. [Google Scholar] [CrossRef]
- Xu, Q.; Hu, M.; Li, M.; Hou, J.; Zhang, X.; Gao, Y.; Chachar, B.; Li, X. Dietary Bioactive Peptide Alanyl-Glutamine Attenuates Dextran Sodium Sulfate-Induced Colitis by Modulating Gut Microbiota. Oxid. Med. Cell Longev. 2021, 2021, 5543003. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Sun, Z.; Zeng, B.; Huang, S.; Zhao, J.; Zhang, Y.; Su, X.; Xu, J.; Wei, H.; Zhang, H. Cow-to-Mouse Fecal Transplantations Suggest Intestinal Microbiome as One Cause of Mastitis. Microbiome 2018, 6, 200. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Hu, M.; Zhang, L.; Gao, Y.; Xu, Q. Dietary Taxifolin Protects Against Dextran Sulfate Sodium-Induced Colitis via NF-ΚB Signaling, Enhancing Intestinal Barrier and Modulating Gut Microbiota. Front. Immunol. 2021, 11, 3915. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, C.; Stojanović, O.; Colin, D.J.; Suarez-Zamorano, N.; Tarallo, V.; Veyrat-Durebex, C.; Rigo, D.; Fabbiano, S.; Stevanović, A.; Hagemann, S.; et al. Gut Microbiota Orchestrates Energy Homeostasis during Cold. Cell 2015, 163, 1360–1374. [Google Scholar] [CrossRef] [Green Version]
- de Menezes, A.B.; Lewis, E.; O’Donovan, M.; O’Neill, B.F.; Clipson, N.; Doyle, E.M. Microbiome Analysis of Dairy Cows Fed Pasture or Total Mixed Ration Diets. FEMS Microbiol. Ecol. 2011, 78, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Meng, Q.; Chen, Y.; Xu, M.; Shen, M.; Gao, R.; Gan, S. Role of Age-Related Shifts in Rumen Bacteria and Methanogens in Methane Production in Cattle. Front. Microbiol. 2017, 8, 1563. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Chou, W.-C.; Lai, Y.; Liang, K.; Tam, J.W.; Brickey, W.J.; Chen, L.; Montgomery, N.D.; Li, X.; Bohannon, L.M.; et al. Multi-Omics Analyses of Radiation Survivors Identify Radioprotective Microbes and Metabolites. Science 2020, 370, eaay9097. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Davenport, E.R.; Waters, J.L.; Clark, A.G.; Ley, R.E. Cross-Species Comparisons of Host Genetic Associations with the Microbiome. Science 2016, 352, 532–535. [Google Scholar] [CrossRef] [Green Version]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment Dominates over Host Genetics in Shaping Human Gut Microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef]
- Oliphant, K.; Allen-Vercoe, E. Macronutrient Metabolism by the Human Gut Microbiome: Major Fermentation by-Products and Their Impact on Host Health. Microbiome 2019, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xu, Y.-Q.; Yuan, Y.-X.; Xu, P.-W.; Zhang, C.; Li, F.; Wang, L.-N.; Yin, C.; Zhang, L.; Cai, X.-C.; et al. Succinate Induces Skeletal Muscle Fiber Remodeling via SUCNR1 Signaling. EMBO Rep. 2021, 22, e53027. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (5’ to 3’) | GenBank No. |
|---|---|---|
| For pigs | ||
| Bcat2 | F: GCCTGAAGGCGTACAAAGG; R: GATGCACTCCAGCAACTCG | |
| Bckdh | F: CCAGATGCCCGTCCACTAC; R: CCCCCTCTCCGAAGTAACAG | |
| Bckdk | F: TCCGACCATGATGCTCTATTC; R: GAAGTCCTT GATGCGGTG G | |
| CoxVa | F: CCGCCGCTATCCAGTCAATTCG; R: ATTTCCTTATGAGGTCCTGCTTTGTCC | XM_005666147.2 |
| CoXVIIb | F: GGCAAGGCAGAACCACCAGAAG; R: TGGGTACACAGTGCTTTTACTTGGC | XM_003135201.3 |
| mCpt-1b | F: CACGGCAACTGCTACAACA; R: CCAGGACGAACTCCCAGA | |
| CytC | F: AACTGGTCAAGCACCTGGTTATAGC; R:ACCAGACGGAGAATCAATGAATGAGTC | XM_003127002.4 |
| GAPDH | F: GTCGGAGTGAACGGATTTGG; R: CAATGTCCACTTTGCCAGAGTTAA | NM_001206359.1 |
| Lcad | F: TGACTGGATTCTCAATGGAAGCAAGG; R: CACTCGCTGGCAACCGTACATC | NM_213897.1 |
| Ldh | F: TCTTGACCTATGTGGCTTGG; R: AGCACTGTCCACCA CCTGTT | XM_013994501.2 |
| Mcad | F: GGCGAGTACCCTGTCCCACTAC; R: GCCTGCTCCTGGTTCTGTTACAC | NM_214039.1 |
| MyHCI | F: CAAGGCAGAGATGGAGCGGAAG; R: CTCATTGCGGCTGCGTGTCT | NM_213855 |
| MyHCIIa | F: GGAAGCTCGCAACGCAGAAGA; R: TCATCCAGACGGTGCTGTAGGT | NM_214136 |
| MyHCIIb | F: CGGAAGAGGCGGAGGAACAATC; R: TGACCTGGGACTCGGCAATGT | NM_001104951 |
| MyHCIIx | F: GTACAATGCGGTGGGTGCTCTG; R: GCTGCTGGTTGATGCGAGTGA | NM_001123141 |
| MyoD | F: CTGCTACGACGGCACCTATT; R: CACGATGCTGGACAGACAG | NM001002824 |
| MyoG | F: AGTGGAGGATGTGGCTGTG; R: AGAAGTGGTGCCGTCTGTG | NM001012406 |
| Pdh | F: ATGAAGAGGGAGGGTGGGAGTTTC; R: GGCGATAGATGGAGTTCCTGTTGTG | XM_003360244.4 |
| Pfk | F: GGCTTTGAGGCTTACACAGG; R: GGATGACCACAAACGGGATG | XM_021091211.1 |
| Pk | F: TCGATGAGATCCTGGAAGCC; R: TCTTCTGAGCCAGGAAGACC | XM_021099125.1 |
| Sdh | F: TGCGAACGGAACCATAAGGACATC; R: GTGCTCCTGGAACGGCTTCTTC | XM_021076931.1 |
| Tfam | F: GCTCCTCCTCCTTCGTCGTAGTC; R: GCACCCGTAGACAAAGCACTGAC | NM_001130211.1 |
| For mice | ||
| Bcat2 | F: GCGTCATCTTGCCTGGAGTAGTTC; R: TTCCTTCATAGTGACCTTGCGTTCTG | |
| Bckdh | F: TGGTGGGATGAGGAACAGGAGAAG; R: TACACATCGGAGAAGAGGAGGCTTG | |
| Bckdk | F: AAGGGAGTAGGAGAAGCAGGAAAGG; GCGTCAAGTGAGGGAACTGGTTAC | |
| CoxVa | F: TTGATGCCTGGGAATTGCGTAAAG; R: AACAACCTCCAAGATGCGAACAG | |
| CoXVIIb | F: TTTCAGGACGCTTTGCAAGG; R: TGCTTCGAACTTGGAGACGG | |
| mCpt-1b | F: ACTCTTGGAAGAAGTTCA; R: AGTATCTTTGACAGCTGGGAC | |
| CytC | F: CCAGTGCCACACTGTGGAAAA; R: TCTCCCCAGGTGATGCCTTT | |
| Gapdh | F: CATCACTGCCACCCAGAAGACTG; R: ATGCCAGTGAGCTTCCCGTTCAG | |
| Lcad | F: GTAGCTTATGAATGTGTGCACTC; R: GTCTTGCGATCAGCTCTTTCATTA | |
| Ldh | F: GGAAGGAGGTTCACAAGCAG; R: TCACAACATCCGAGATTCCA | |
| Mcad | F: GATCGCAATGGGTGCTTTTGATAGA A; R: AGCTGATTGGCAATGTCTCCAGCAAA | |
| MyHCI | F: CCAAGGGCCTGAATGAGGAG; R: GCAAAGGCTCCAGGTCTGAG | |
| MyHCIIa | F: AAGCGAAGAGTAAGGCTGTC; R: GTGATTGCTTGCAAAGGAAC | |
| MyHCIIb | F: ACAAGCTGCGGGTGAAGAGC; R: CAGGACAGTGACAAAGAACG | |
| MyHCIIx | F: CCAAGTGCAGGAAAGTGACC; R: AGGAAGAGACTGACGAGCTC | |
| MyoD | F: GAGGATCCGATGGAGCTTCTATCG; R: CGGATCCTCTCAAAGCACCTGATA | |
| MyoG | F: TTTGCAGTGGATCTTGGGAACCTTC; R: GTCAGACGGCAGCTTTACAAACAAC | |
| Pdh | F: GAAGGCCCTGCATTCAACTTC; R: ATAGGGACATCAGCACCAGTGA | |
| Pfk | F: CAGTCAGTGCCAACATAACCAA; R: CGGGATGCAGAGCTCATCA | |
| Pk | F: TGCCGTGCTGAATGCCTGGG; R: CGCCACCCGGTCAGCACAAT | |
| Sdh | F: GAAAGGCGGGCAGGCTCATC; R: CACCACGGCACTCCCCATTTT | |
| Tfam | F: GCTCTACACGCCCCTGGTTTCTGG; R: TCGCTGTAGTGCCTGCTGCTCCTG |
| Indices | Results | |||||||
|---|---|---|---|---|---|---|---|---|
| Pigs | Mice | |||||||
| EH | LY | SEM | p | CG | AG | SEM | p | |
| Chao 1 | 842.6 a | 765.3 b | 25.81 | 0.200 | 497.7 a | 591.7 b | 12.77 | 0.007 |
| Simpson | 0.02 a | 0.04 b | 0.004 | 0.004 | 0.07 a | 0.03 b | 0.01 | 0.016 |
| Shannon | 4.96 a | 4.58 b | 0.07 | 0.004 | 3.89 a | 4.41 b | 0.08 | 0.023 |
| Good’s Coverage | >99% | >99% | >99% | >99% | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, L.; Li, M.; Zhou, S.; Zhu, X.; Zhang, X.; Xu, Q. Trans-Species Fecal Transplant Revealed the Role of the Gut Microbiome as a Contributor to Energy Metabolism and Development of Skeletal Muscle. Metabolites 2022, 12, 769. https://doi.org/10.3390/metabo12080769
Cai L, Li M, Zhou S, Zhu X, Zhang X, Xu Q. Trans-Species Fecal Transplant Revealed the Role of the Gut Microbiome as a Contributor to Energy Metabolism and Development of Skeletal Muscle. Metabolites. 2022; 12(8):769. https://doi.org/10.3390/metabo12080769
Chicago/Turabian StyleCai, Liyuan, Min Li, Shuyi Zhou, Xiaoyan Zhu, Xianghua Zhang, and Qingbiao Xu. 2022. "Trans-Species Fecal Transplant Revealed the Role of the Gut Microbiome as a Contributor to Energy Metabolism and Development of Skeletal Muscle" Metabolites 12, no. 8: 769. https://doi.org/10.3390/metabo12080769
APA StyleCai, L., Li, M., Zhou, S., Zhu, X., Zhang, X., & Xu, Q. (2022). Trans-Species Fecal Transplant Revealed the Role of the Gut Microbiome as a Contributor to Energy Metabolism and Development of Skeletal Muscle. Metabolites, 12(8), 769. https://doi.org/10.3390/metabo12080769