
Grape Cultivar Features Differentiate the Grape Rhizosphere Microbiota

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Richness and Diversity of Microbiome Communities Associated with the Root Systems of Different Grape Varieties
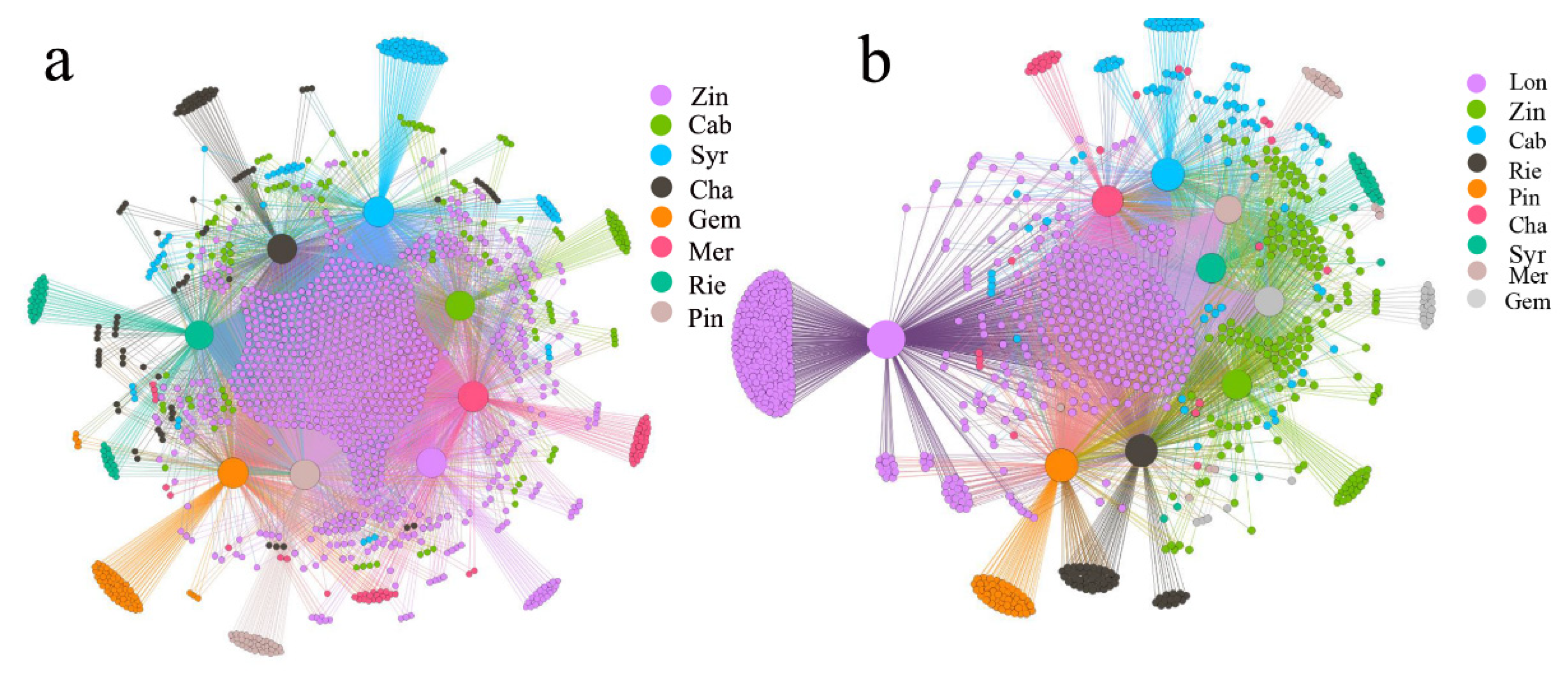
2.2. Grape Microbiome Distribution in the Rhizosphere of Different Varieties of Wine Grape
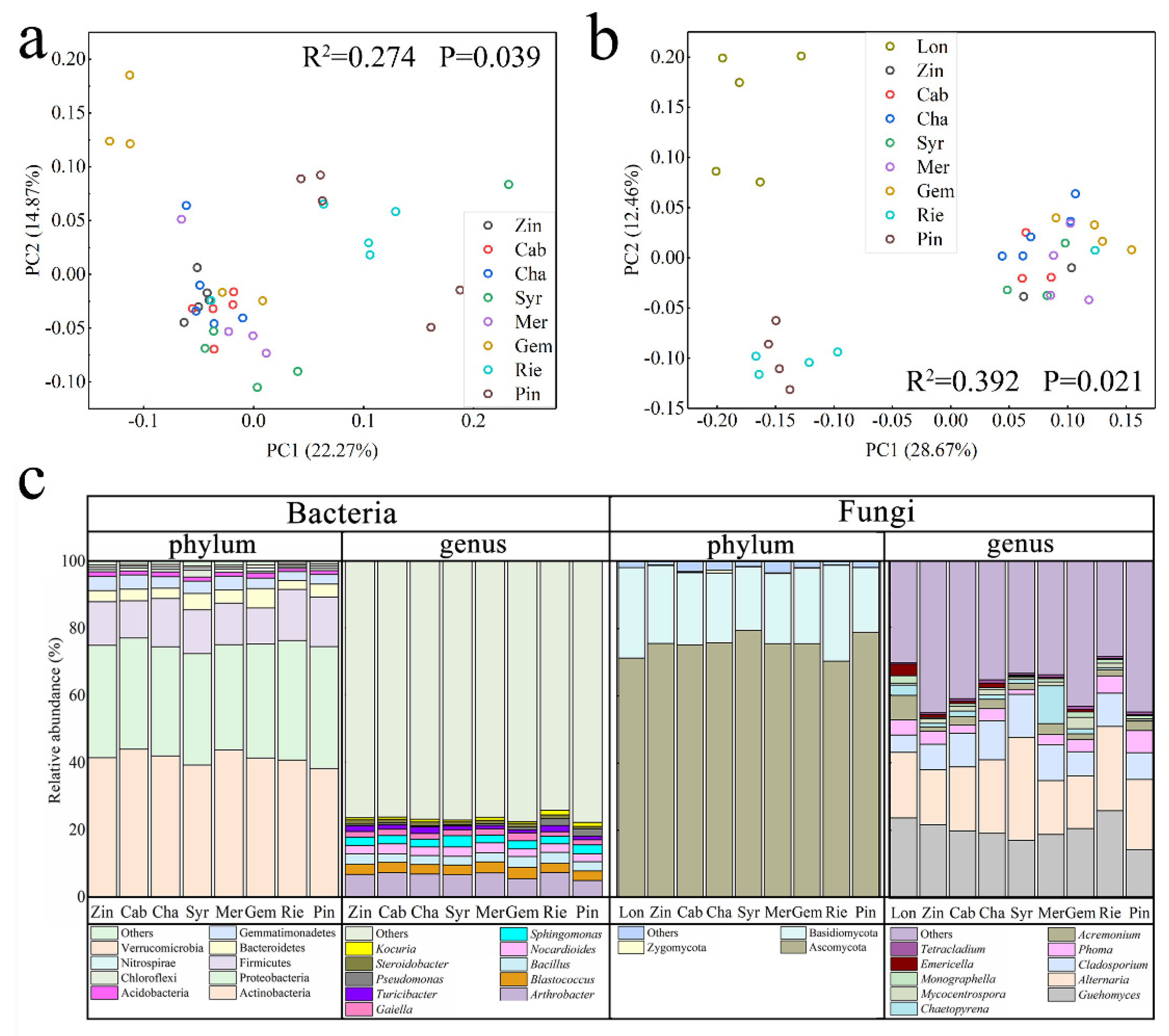
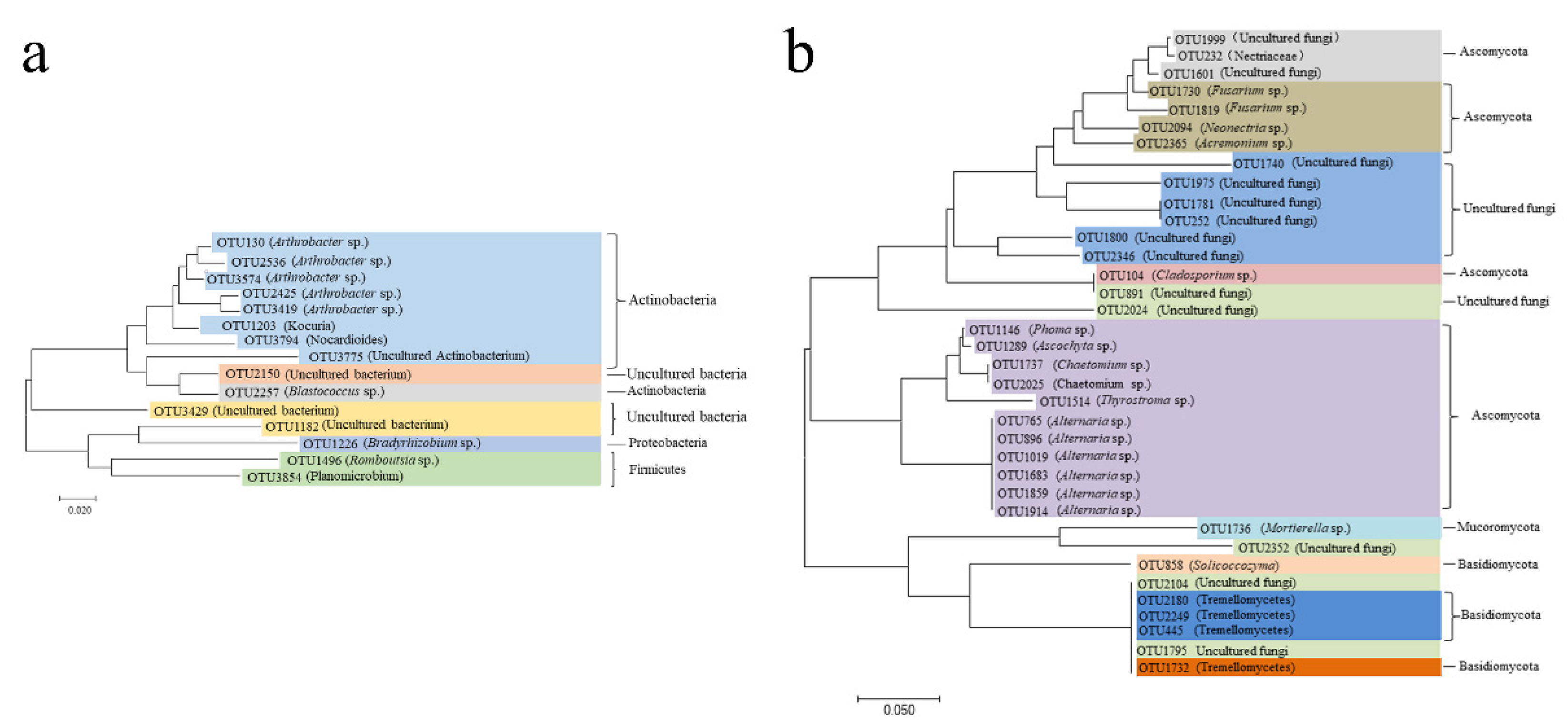
2.3. Differences in Bacterial and Fungal Diversity among Varieties
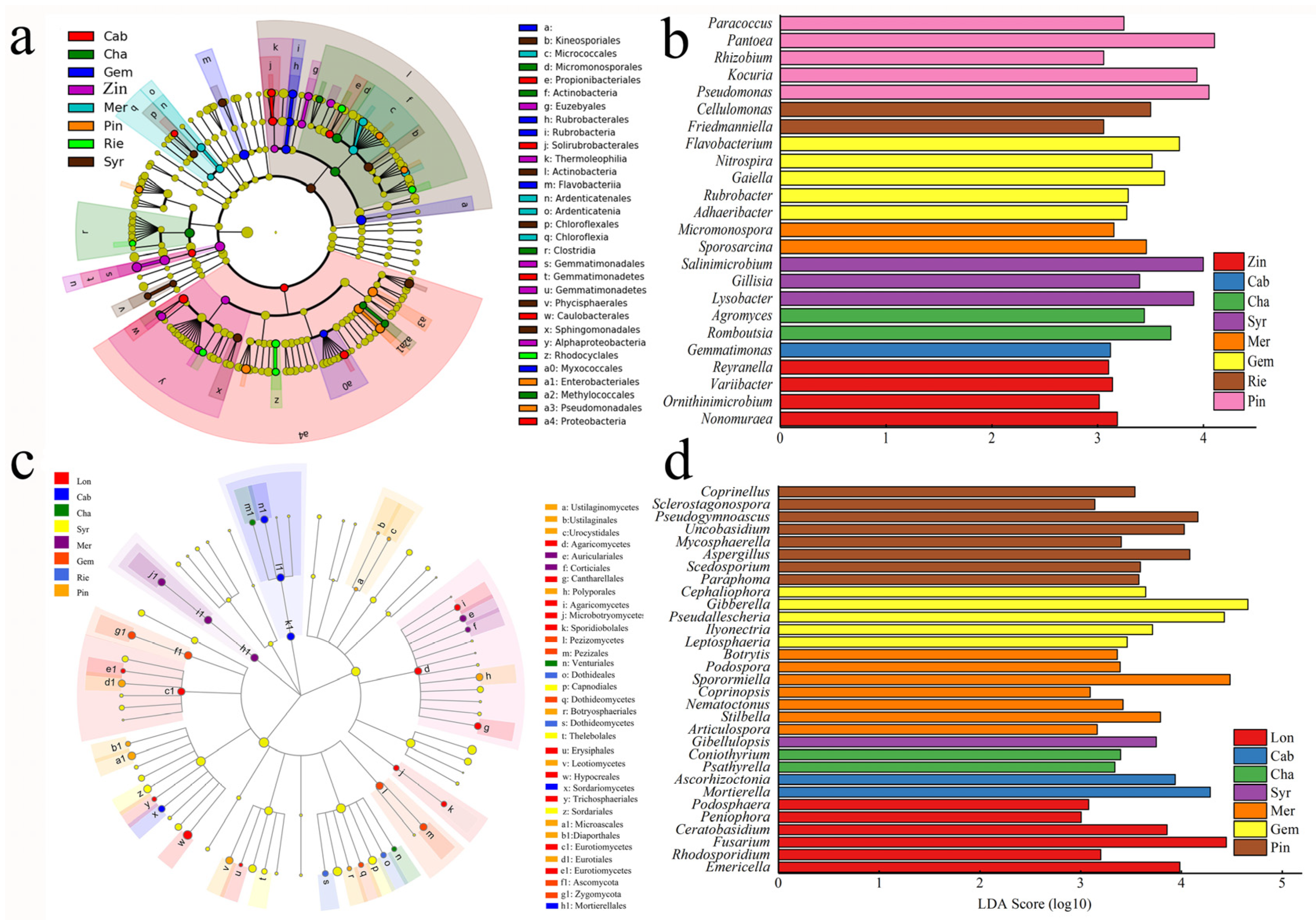
2.4. Functional Genomics of Grapevine Rhizosphere Community
3. Discussion
4. Materials and Methods
4.1. Site Description and Sample Collection
4.2. DNA Extraction and Sequencing
4.3. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hartmann, A.; Rothballer, M.; Schmid, M. Lorenz Hiltner, a pioneer in rhizosphere microbial ecology and soil bacteriology research. Plant Soil 2008, 312, 7–14. [Google Scholar] [CrossRef]
- Uroz, S.; Calvaruso, C.; Turpault, M.P.; Pierrat, J.C.; Mustin, C.; Frey-Klett, P. Effect of the Mycorrhizosphere on the Genotypic and Metabolic Diversity of the Bacterial Communities Involved in Mineral Weathering in a Forest Soil. Appl. Environ. Microbiol. 2007, 73, 3019–3027. [Google Scholar] [CrossRef] [Green Version]
- Jacoby, R.; Peukert, M.; Succurro, A.; Koprivova, A.; Kopriva, S. The Role of Soil Microorganisms in Plant Mineral Nutrition-Current Knowledge and Future Directions. Front. Plant Sci. 2017, 8, 1617. [Google Scholar] [CrossRef] [Green Version]
- Smercina, D.N.; Evans, S.E.; Friesen, M.L.; Tiemann, L.K. To fix or not to fix: Controls on free-living nitrogen fixation in the rhizosphere. Appl. Environ. Microbiol. 2019, 85, e02546-18. [Google Scholar] [CrossRef] [Green Version]
- Steenhoudt, O.; Vanderleyden, J. Azospirillum, a free-living nitrogen-fixing bacterium closely associated with grasses: Genetic, biochemical and ecological aspects. FEMS Microbiol. Rev. 2000, 24, 487–506. [Google Scholar] [CrossRef]
- Long, S.R. Rhizobium symbiosis: Nod factors in perspective. Plant Cell 1996, 8, 1885–1898. [Google Scholar]
- Sprent, J.I.; James, E.K. Legume evolution: Where do nodules and mycorrhizas fit in? Plant Physiol. 2007, 144, 575–581. [Google Scholar] [CrossRef] [Green Version]
- Mus, F.; Crook, M.B.; Garcia, K.; Garcia Costas, A.; Geddes, B.A.; Kouri, E.D.; Paramasivan, P.; Ryu, M.H.; Oldroyd, G.; Poole, P.S.; et al. Symbiotic Nitrogen Fixation and the Challenges to Its Extension to Nonlegumes. Appl. Environ. Microbiol. 2016, 82, 3698–3710. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, A.M. Developmental biology of legume nodulation. New Phytol. 1992, 122, 211–237. [Google Scholar] [CrossRef]
- Hayat, R.; Ali, S.; Amara, U.; Khalid, R.; Ahmed, I. Soil beneficial bacteria and their role in plant growth promotion: A review. Ann. Microbiol. 2010, 60, 579–598. [Google Scholar] [CrossRef]
- Jadhav, H.P.; Shaikh, S.S.; Sayyed, R.Z. Role of Hydrolytic Enzymes of Rhizoflora in Biocontrol of Fungal Phytopathogens: An Overview. In Rhizotrophs: Plant Growth Promotion to Bioremediation; Mehnaz, S., Ed.; Microorganisms for Sustainability; Springer: Singapore, 2017; Volume 2, pp. 183–203. [Google Scholar]
- Kristin, A.; Miranda, H. The root microbiota—A fingerprint in the soil? Plant Soil 2013, 370, 671–686. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A.H.M. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef]
- Aira, M.; Gómez-Brandón, M.; Lazcano, C.; Bååth, E.; Domínguez, J. Plant genotype strongly modifies the structure and growth of maize rhizosphere microbial communities. Soil Biol. Biochem. 2015, 42, 2276–2281. [Google Scholar] [CrossRef]
- Sugiyama, A.; Ueda, Y.; Takase, H.; Yazaki, K. Pyrosequencing assessment of rhizosphere fungal communities from a soybean field. Can. J. Microbiol. 2014, 60, 687–690. [Google Scholar] [CrossRef]
- Haichar, F.E.Z.; Marol, C.; Berge, O.; Rangel-Castro, J.I.; Prosser, J.I.; Balesdent, J.m.; Heulin, T.; Achouak, W. Plant host habitat and root exudates shape soil bacterial community structure. ISME J. 2008, 2, 1221–1230. [Google Scholar] [CrossRef]
- Knight, S.; Klaere, S.; Fedrizzi, B.; Goddard, M.R. Regional microbial signatures positively correlate with differential wine phenotypes: Evidence for a microbial aspect to terroir. Sci. Rep. 2015, 5, 14233. [Google Scholar] [CrossRef] [Green Version]
- Martins, G.; Lauga, B.; Miot-Sertier, C.; Mercier, A.; Lonvaud, A.; Soulas, M.L.; Soulas, G.; Masneuf-Pomarède, I. Characterization of Epiphytic Bacterial Communities from Grapes, Leaves, Bark and Soil of Grapevine Plants Grown, and Their Relations. PLoS ONE 2013, 8, e73013. [Google Scholar] [CrossRef] [Green Version]
- Zarraonaindia, I.; Owens, S.M.; Weisenhorn, P.; West, K.; Hampton-Marcell, J.; Lax, S.; Bokulich, N.A.; Mills, D.A.; Martin, G.; Taghavi, S. The Soil Microbiome Influences Grapevine-Associated Microbiota. mBio 2015, 6, e02527-14. [Google Scholar] [CrossRef] [Green Version]
- Sakuradani, E.; Shimizu, S. Single cell oil production by Mortierella alpina. J. Biotechnol. 2009, 144, 31–36. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Collins, T.S.; Masarweh, C.; Allen, G.; Heymann, H.; Ebeler, S.E.; Mills, D.A. Associations among Wine Grape Microbiome, Metabolome, and Fermentation Behavior Suggest Microbial Contribution to Regional Wine Characteristics. mBio 2016, 7, e00631-16. [Google Scholar] [CrossRef] [Green Version]
- Yergeau, E.; Bezemer, T.M.; Hedlund, K.; Mortimer, S.R.; Kowalchuk, G.A.; van der Putten, W.H. Influences of space, soil, nematodes and plants on microbial community composition of chalk grassland soils. Environ. Microbiol. 2010, 12, 2096–2106. [Google Scholar] [CrossRef]
- Sabate, J.; Cano, J.; Esteve-Zarzoso, B.; Guillamón, J.M. Isolation and identification of yeasts associated with vineyard and winery by RFLP analysis of ribosomal genes and mitochondrial DNA. Microbiol. Res. 2002, 157, 267–274. [Google Scholar] [CrossRef]
- Manoharan, L.; Rosenstock, N.P.; Williams, A.; Hedlund, K. Agricultural management practices influence AMF diversity and community composition with cascading effects on plant productivity. Appl. Soil Ecol. 2017, 115, 53–59. [Google Scholar] [CrossRef]
- Marasco, R.; Rolli, E.; Fusi, M.; Michoud, G.; Daffonchio, D. Grapevine rootstocks shape underground bacterial microbiome and networking but not potential functionality. Microbiome 2018, 6, 3. [Google Scholar] [CrossRef]
- Wallenstein, M.D.; Hall, E.K. A trait-based framework for predicting when and where microbial adaptation to climate change will affect ecosystem functioning. Biogeochemistry 2012, 109, 35–47. [Google Scholar] [CrossRef]
- Weinert, N.; Piceno, Y.; Ding, G.C.; Meincke, R.; Heuer, H.; Berg, G.; Schloter, M.; Andersen, G.; Smalla, K. PhyloChip hybridization uncovered an enormous bacterial diversity in the rhizosphere of different potato cultivars: Many common and few cultivar-dependent taxa. FEMS Microbiol. Ecol. 2011, 75, 497–506. [Google Scholar] [CrossRef]
- Martiny, J.B.H.; Bohannan, B.J.M.; Brown, J.H.; Colwell, R.K.; Fuhrman, J.A.; Green, J.L.; Horner-Devine, M.C.; Kane, M.; Krumins, J.A.; Kuske, C.R. Microbial biogeography: Putting microorganisms on the map. Nat. Rev. Microbiol. 2006, 4, 102–112. [Google Scholar] [CrossRef]
- Coller, E.; Cestaro, A.; Zanzotti, R.; Bertoldi, D.; Pindo, M.; Larger, S.; Albanese, D.; Mescalchin, E.; Donati, C. Microbiome of vineyard soils is shaped by geography and management. Microbiome 2019, 7, 140. [Google Scholar] [CrossRef] [Green Version]
- Siddiqi, M.Z.; Kim, Y.-J.; Hoang, V.-A.; Siddiqi, M.H.; Huq, M.A.; Yang, D.-C. Arthrobacter ginsengisoli sp. nov., isolated from soil of a ginseng field. Arch. Microbiol. 2014, 196, 863–870. [Google Scholar] [CrossRef]
- Hoang, V.-A.; Kim, Y.-J.; Nguyen, N.-L.; Yang, D.-C. Arthrobacter gyeryongensis sp. nov., isolated from soil of a Gynostemma pentaphyllum field. Int. J. Syst. Evol. Microbiol. 2014, 64, 420–425. [Google Scholar] [CrossRef] [Green Version]
- Sacks, L.E. Observations on the morphogenesis of Arthrobacter citreus, spec nov. J. Bacteriol. 1954, 67, 342–345. [Google Scholar] [CrossRef] [Green Version]
- Ensign, J.C.; Rittenberg, S.C. A crystalline pigment produced from 2-hydroxypyridine by Arthrobacter crystallopoietes n. sp. Arch. Mikrobiol. 1963, 47, 137–153. [Google Scholar] [CrossRef]
- Salazar, O.; Valverde, A.; Genilloud, O. Real-time PCR for the detection and quantification of geodermatophilaceae from stone samples and identification of new members of the genus blastococcus. Appl. Environ. Microbiol. 2006, 72, 346–352. [Google Scholar] [CrossRef] [Green Version]
- Sghaier, H.; Hezbri, Z.; Ghodhbane-Gtari, F.; Pujic, P.; Sen, A.; Daffonchio, D.; Boudabous, A.; Tisa, L.S.; Klenk, H.P.; Armengaud, J.; et al. Stone-dwelling actinobacteria Blastococcus saxobsidens, Modestobacter marinus and Geodermatophilus obscurus proteogenomes. ISME J. 2016, 10, 21–29. [Google Scholar] [CrossRef]
- Perazzolli, M.; Antonielli, L.; Storari, M.; Puopolo, G.; Pertot, I. Resilience of the Natural Phyllosphere Microbiota of the Grapevine to Chemical and Biological Pesticides. Appl. Environ. Microbiol. 2014, 80, 3585–3596. [Google Scholar] [CrossRef] [Green Version]
- Motta, S.D.; Soares, L.M.V. Survey of Brazilian tomato products for alternariol, alternariol monomethyl ether, tenuazonic acid and cyclopiazonic acid. Food Addit. Contam. 2001, 18, 630–634. [Google Scholar] [CrossRef]
- Fredj, S.M.B.; Chebil, S.; Lebrihi, A.; Lasram, S.; Ghorbel, A.; Mliki, A. Occurrence of pathogenic fungal species in Tunisian vineyards. Int. J. Food Microbiol. 2007, 113, 245–250. [Google Scholar] [CrossRef]
- Fleet, G.H. Yeast interactions and wine flavour. Int. J. Food Microbiol. 2003, 86, 11–22. [Google Scholar] [CrossRef]
- Park, J.Y.; Han, S.H.; Lee, J.H.; Han, Y.S.; Lee, Y.S.; Rong, X.; McSpadden Gardener, B.B.; Park, H.-S.; Kim, Y.C. Draft Genome Sequence of the Biocontrol Bacterium Pseudomonas putida B001, an Oligotrophic Bacterium That Induces Systemic Resistance to Plant Diseases. J. Bacteriol. 2011, 193, 6795–6796. [Google Scholar] [CrossRef] [Green Version]
- Hirano, S.S.; Upper, C.D. Bacteria in the Leaf Ecosystem with Emphasis on Pseudomonas syringae—A Pathogen, Ice Nucleus, and Epiphyte. Microbiol. Mol. Biol. Rev. 2000, 64, 624–653. [Google Scholar] [CrossRef] [Green Version]
- Luginbuehl, L.H.; Menard, G.N.; Kurup, S. Fatty acids in arbuscular mycorrhizal fungi are synthesized by the host plant. Science 2017, 356, 1175. [Google Scholar] [CrossRef] [Green Version]
- Wang, E. Plants transfer lipids to sustain colonization by mutualistic mycorrhizal and parasitic fungi. Science 2017, 356, 1172. [Google Scholar]
- Zhang, S.; Xi, C.; Zhong, Q.; Huang, Z.; Bai, Z. Relations among epiphytic microbial communities from soil, leaves and grapes of the grapevine. Front. Life Sci. 2017, 10, 73–83. [Google Scholar] [CrossRef]
- Fung, R.W.M.; Gonzalo, M.; Fekete, C.; Kovacs, L.G.; He, Y. Powdery Mildew Induces Defense-Oriented Reprogramming of the Transcriptome in a Susceptible but not in a Resistant Grapevine. Plant Physiol. 2008, 146, 236–249. [Google Scholar] [CrossRef] [Green Version]
- Marzano, M.; Fosso, B.; Manzari, C.; Grieco, F.; Intranuovo, M.; Cozzi, G.; Mulè, G.; Scioscia, G.; Valiente, G.; Tullo, A.; et al. Complexity and Dynamics of the Winemaking Bacterial Communities in Berries, Musts, and Wines from Apulian Grape Cultivars through Time and Space. PLoS ONE 2016, 11, e0157383. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Thorngate, J.H.; Richardson, P.M.; Mills, D.A. Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc. Natl. Acad. Sci. USA 2014, 111, E139–E148. [Google Scholar] [CrossRef] [Green Version]
- Mendes, L.W.; Kuramae, E.E.; Navarrete, A.A.; van Veen, J.A.; Tsai, S.M. Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J. 2014, 8, 1577–1587. [Google Scholar] [CrossRef]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [Green Version]
- Chelius, M.K.; Triplett, E.W. The Diversity of Archaea and Bacteria in Association with the Roots of Zea mays L. Microb. Ecol. 2001, 41, 252–263. [Google Scholar] [CrossRef]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes - application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef]
- Bodenhausen, N.; Horton, M.W.; Bergelson, J. Bacterial communities associated with the leaves and the roots of Arabidopsis thaliana. PLoS ONE 2013, 8, e56329. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gever, D. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.; Van Treuren, W.; Lozupone, C.; Faust, K.; Friedman, J.; Deng, Y.; Xia, L.C.; Xu, Z.Z.; Ursell, L.; Alm, E.J. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 2016, 10, 1669–1681. [Google Scholar] [CrossRef]
- Faust, K.; Sathirapongsasuti, J.F.; Izard, J.; Segata, N.; Gevers, D. Microbial Co-occurrence Relationships in the Human Microbiome. PLoS Comput. Biol. 2012, 8, e1002606. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coverage | ACE (103) | Chao1 (103) | Simpson (10−2) | Shannon | |
|---|---|---|---|---|---|
| Zin | 0.958 ± 0.018 a | 2.12 ± 0.167 ab | 2.02 ± 0.264 ab | 0.593 ± 0. 04 a | 6.09 ± 0.098 a |
| Cab | 0.960 ± 0.016 a | 2.16 ± 0.158 ab | 2.05 ± 0.131 ab | 0. 617 ± 0.07 a | 6.07 ± 0.069 a |
| Cha | 0.967 ± 0.007 a | 2.03 ± 0.165 ab | 2.04 ± 0.164 ab | 0.646 ± 0.05 a | 6.05 ± 0.071 a |
| Syr | 0.972 ± 0.011 a | 2.06 ± 0.163 ab | 2.07 ± 0.151 ab | 0.693 ± 0.05 a | 5.99 ± 0.083 a |
| Mer | 0.959 ± 0.005 a | 1.89 ± 0.110 a | 1.87 ± 0.128 a | 0.678 ± 0.06 a | 6.02 ± 0.080 a |
| Gem | 0.956 ± 0.013 a | 2.02 ± 0.154 ab | 1.89 ± 0.162 a | 0.658 ± 0.16 a | 6.05 ± 0.123 a |
| Rie | 0.966 ± 0.007 a | 2.07 ± 0.137 ab | 2.04 ± 0.146 ab | 0.677 ± 0.11 a | 5.99 ± 0.119 a |
| Pin | 0.978 ± 0.005 a | 2.25 ± 0.181 b | 2.29 ± 0.185 b | 0.789 ± 0.47 a | 5.99 ± 0.234 a |
| Coverage | ACE (102) | Chao1 (102) | Simpson (10−2) | Shannon | |
|---|---|---|---|---|---|
| Zin | 0.997 ± 0.001 a | 9.63 ± 0.506 b | 9.76 ± 0.771 b | 3.64 ± 0.907 a | 4.14 ± 0.119 c |
| Cab | 0.999 ± 0.001 a | 7.28 ± 0.797 a | 7.54 ± 0.638 ab | 7.78 ± 2.76 ab | 3.57 ± 0.301 ab |
| Cha | 0.997 ± 0.001 a | 7.17 ± 0.834 a | 7.09 ± 0.848 a | 4.68 ± 0.956 ab | 4.02 ± 0.128 bc |
| Syr | 0.998 ± 0.001 a | 8.56 ± 0.305 ab | 8.77 ± 0.611 ab | 6.21 ± 2.79 ab | 3.87 ± 0.265a bc |
| Mer | 0.998 ± 0.001 a | 8.23 ± 0.837 ab | 8.23 ± 0.957 ab | 9.80 ± 4.51 b | 3.49 ± 0.339 a |
| Gem | 0.998 ± 0.001 a | 7.30 ± 0.628 a | 7.40 ± 0.669 a | 4.84 ± 1.96 ab | 3.88 ± 0.250 abc |
| Rie | 0.998 ± 0.001 a | 7.66 ± 0.989 a | 7.83 ± 0.975 ab | 5.64 ± 0.963 ab | 3.80 ± 0.126 abc |
| Pin | 0.996 ± 0.003 a | 8.33 ± 1.47 ab | 8.12 ± 1.63 ab | 8.97 ± 2.95 ab | 3.57 ± 0.181 ab |
| Lon | 0.998 ± 0.001 a | 9.13 ± 0.503 ab | 9.06 ± 0.525 ab | 4.74 ± 0.313 ab | 3.97 ± 0.117 abc |
| Bray–Curtis | Unweighted Unifrac | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ADONIS | ANOSIM | ADONIS | ANOSIM | ||||||
| Group | Factor | R2 | p | R | p | R2 | p | R | p |
| Eight | Variety | 0.319 | 0.001 | 0.317 | 0.001 | 0.215 | 0.001 | 0.245 | 0.001 |
| Bray–Curtis | Unweighted Unifrac | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ADONIS | ANOSIM | ADONIS | ANOSIM | ||||||
| Group | Factor | R2 | p | R | p | R2 | p | R | p |
| Nine | Variety | 0.373 | 0.001 | 0.295 | 0.001 | 0.442 | 0.001 | 0.401 | 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, L.; Sun, B.; Wei, Y.; Xu, N.; Zhang, S.; Gu, L.; Bai, Z. Grape Cultivar Features Differentiate the Grape Rhizosphere Microbiota. Plants 2022, 11, 1111. https://doi.org/10.3390/plants11091111
Bao L, Sun B, Wei Y, Xu N, Zhang S, Gu L, Bai Z. Grape Cultivar Features Differentiate the Grape Rhizosphere Microbiota. Plants. 2022; 11(9):1111. https://doi.org/10.3390/plants11091111
Chicago/Turabian StyleBao, Lijun, Bo Sun, Yingxue Wei, Nan Xu, Shiwei Zhang, Likun Gu, and Zhihui Bai. 2022. "Grape Cultivar Features Differentiate the Grape Rhizosphere Microbiota" Plants 11, no. 9: 1111. https://doi.org/10.3390/plants11091111
APA StyleBao, L., Sun, B., Wei, Y., Xu, N., Zhang, S., Gu, L., & Bai, Z. (2022). Grape Cultivar Features Differentiate the Grape Rhizosphere Microbiota. Plants, 11(9), 1111. https://doi.org/10.3390/plants11091111