One-Pot Synthesis of Heavier Group 14 N-Heterocyclic Carbene Using Organosilicon Reductant


Abstract
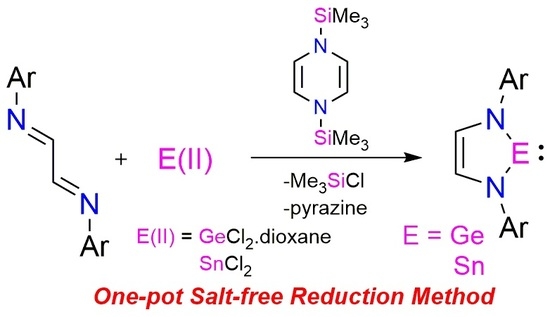
:
{kind=link}
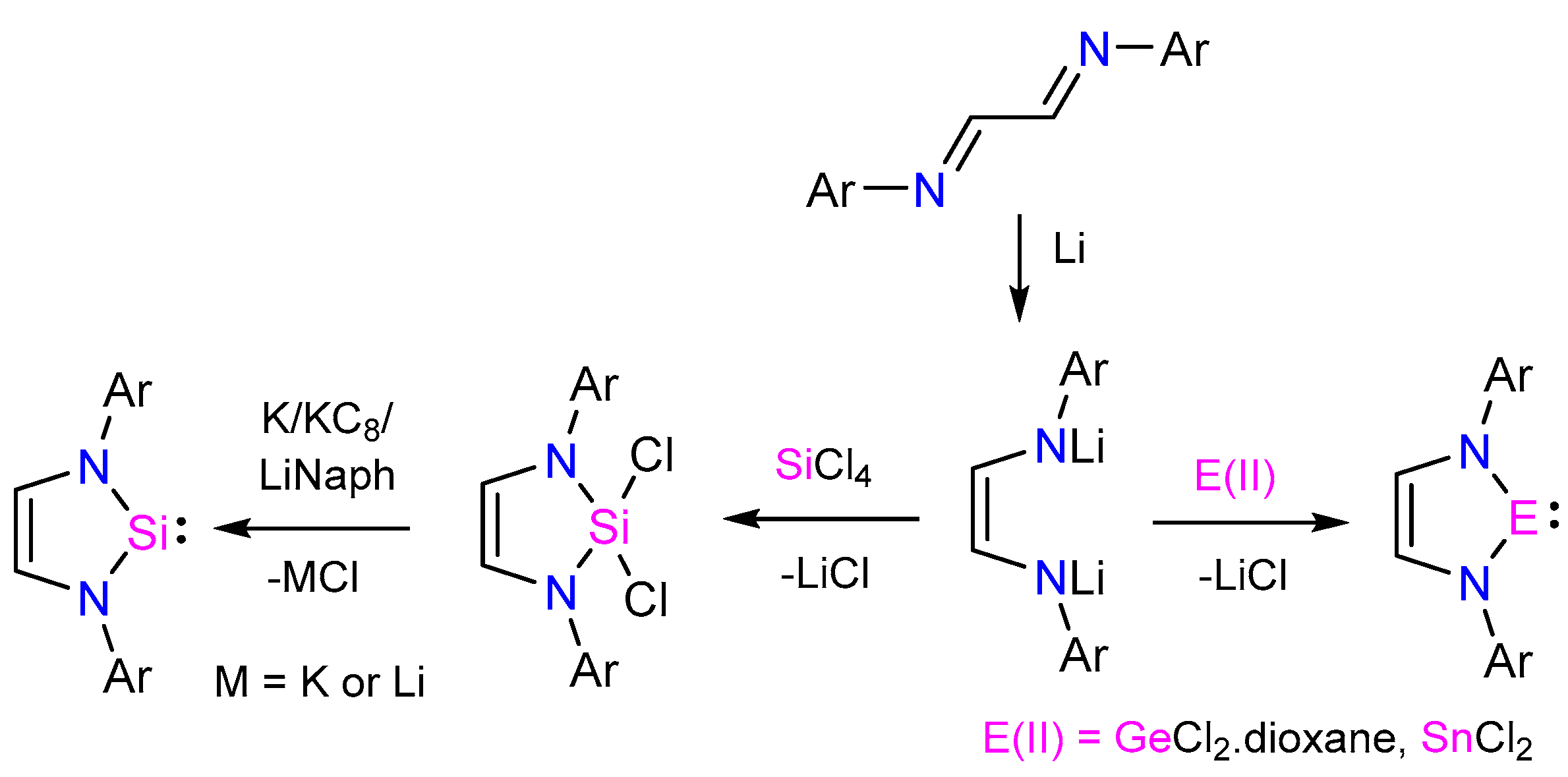{kind=link}
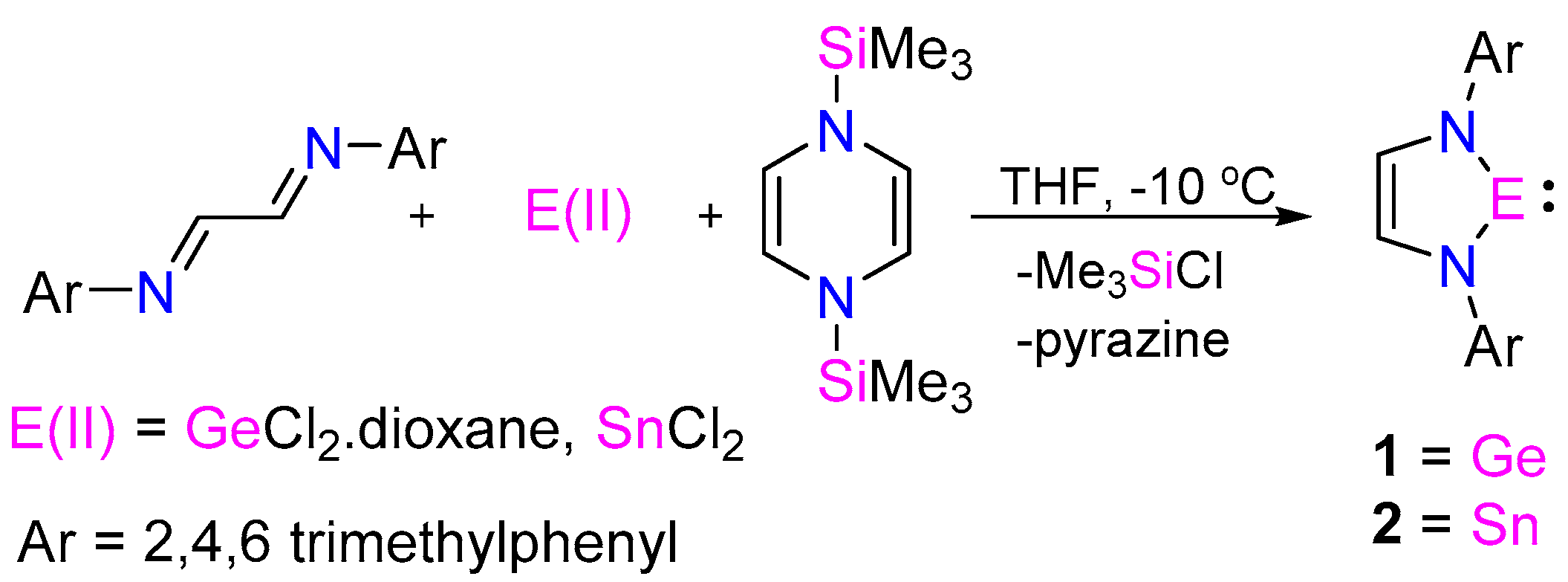{kind=link}
{kind=link}
1. Introduction
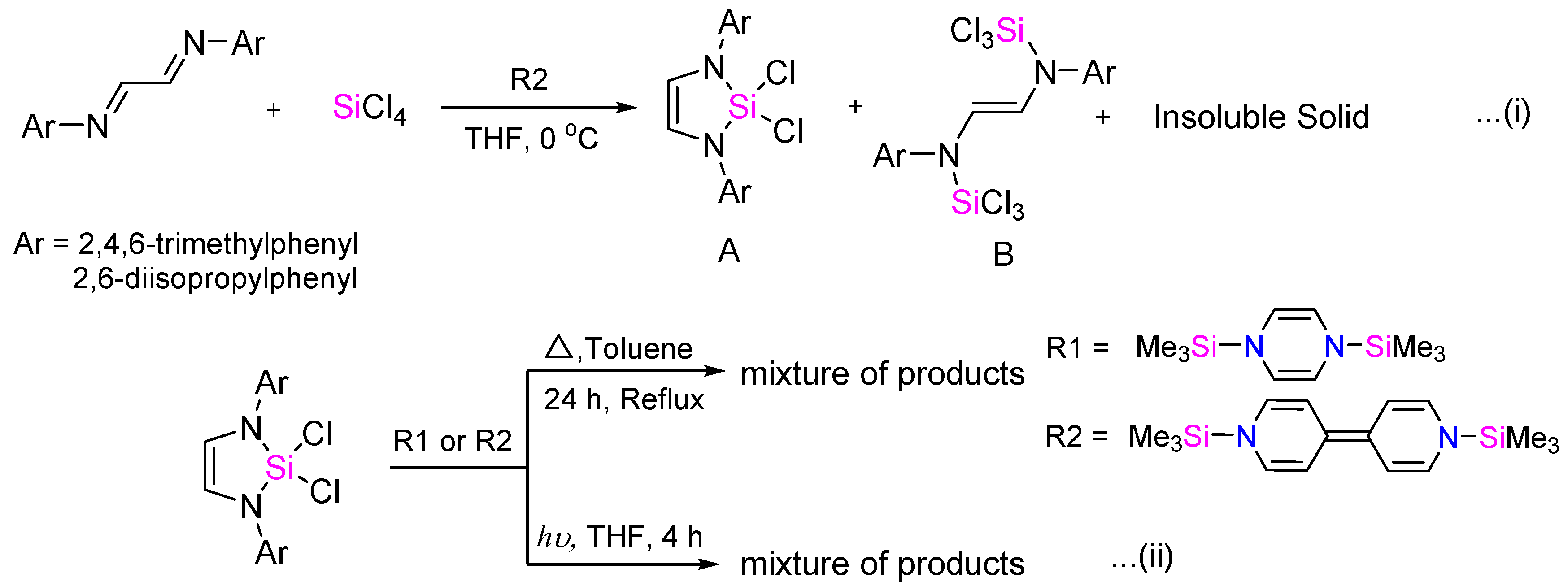
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Experimental Detail
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Arduengo, A.J., III; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Peris, E. Smart N-heterocyclic carbene ligands in catalysis. Chem. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Asay, M.; Jones, C.; Driess, M. N-Heterocyclic carbene analogues with low-valent group 13 and group 14 elements: Syntheses, structures, and reactivities of a new generation of multitalented ligands. Chem. Rev. 2011, 111, 354–396. [Google Scholar] [CrossRef] [PubMed]
- Mizuhata, Y.; Sasamori, T.; Tokitoh, N. Stable Heavier Carbene Analogues. Chem. Rev. 2009, 109, 3479–3511. [Google Scholar] [CrossRef] [PubMed]
- Raoufmoghaddam, S.; Zhou, Y.-P.; Wang, Y.; Driess, M. N-Heterocyclic Silylenes as powerful steering ligands in catalysis. J. Organomet. Chem. 2017, 829, 2–10. [Google Scholar] [CrossRef]
- Blom, B.; Gallego, D.; Driess, M. N-Heterocyclic Silylene Complexes in Catalysis: New frontiers in an emerging field. Inorg. Chem. Front. 2014, 1, 134–148. [Google Scholar] [CrossRef]
- Blom, B.; Stoelzel, M.; Driess, M. New Vistas in N-Heterocyclic Silylene (NHSi) Transition-Metal Coordination Chemistry: Syntheses, Structures and Reactivity towards Activation of Small Molecules. Chem. Eur. J. 2013, 19, 40–62. [Google Scholar] [CrossRef] [PubMed]
- Shoda, S.-I.; Iwata, S.; Yajima, K.; Yagi, K.; Ohnishi, Y.; Kobayashi, S. Synthesis of germanium enolate polymers from germylene monomers. Tetrahedron 1997, 53, 15281–15295. [Google Scholar] [CrossRef]
- Shoda, S.-I.; Iwata, S.; Kim, H.J.; Hiraishi, M.; Kobayashi, S. Poly(germanium thiolate): A new class of organometallic polymers having a germanium-sulfur bond in the main chain. Macromol. Chem. Phys. 1996, 197, 2437–2445. [Google Scholar] [CrossRef]
- Kobayashi, S.; Iwata, S.; Hiraishi, M. Novel 2:1 periodic copolymers from cyclic germylenes and p-benzoquinone derivatives. J. Am. Chem. Soc. 1994, 116, 6047–6048. [Google Scholar] [CrossRef]
- Veprek, S.; Prokop, J.; Glatz, F.; Merica, R.; Klingan, F.R.; Herrmann, W.A. Organometallic chemical vapour deposition of germanium from a cyclic germylene, 1,3-Di-tert-butyl-1,3,2-diazagermolidin-2-ylidene. Chem. Mater. 1996, 8, 825–831. [Google Scholar] [CrossRef]
- Hermann, W.A.; Denk, M.; Behm, J.; Scherer, W.; Klingan, F.; Bock, H.; Solouki, B.; Wagner, M. Stable cyclic germanediyls (“cyclogermylenes”): Synthesis, structure, metal complexes and thermolysis. Angew. Chem. Int. Ed. 1992, 31, 1485–1488. [Google Scholar] [CrossRef]
- Baker, R.J.; Jones, C.; Mills, D.P.; Pierce, G.A.; Waugh, M. Investigation into the preparation of groups 13-15 N-heterocyclic carbene analogues. Inorg. Chim. Acta 2008, 361, 427–435. [Google Scholar] [CrossRef]
- Piskunov, A.V.; Aivaz’yan, I.A.; Cherkasov, V.K.; Abakumov, G.A. New paramagnetic N-heterocyclic stannylenes: An EPR study. J. Organomet. Chem. 2006, 691, 1531–1534. [Google Scholar] [CrossRef]
- Veith, M. Cyclic nitrogen derivatives of tetra- and divalent tin. Angew. Chem. Int. Ed. 1975, 14, 263–264. [Google Scholar] [CrossRef]
- Park, P.; Schäfer, A.; Mitra, A.; Haase, D.; Saak, W.; West, R.; Müller, T. Synthesis and reactivity of N-aryl substituted N-heterocyclic silylenes. J. Organomet. Chem. 2010, 695, 398–408. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, J.; Song, H.; Cui, C. N-Aryl substituted heterocyclic silylenes. Dalton Trans. 2009, 5444–5446. [Google Scholar] [CrossRef] [PubMed]
- Denk, M.; Lennon, R.; Hayashi, R.; West, R.; Belyakov, A.V.; Verne, H.P.; Haaland, A.; Wagner, M.; Metzler, N. Synthesis and structure of a stable silylene. J. Am. Chem. Soc. 1994, 116, 2691–2692. [Google Scholar] [CrossRef]
- Saito, T.; Nishiyama, H.; Tanahashi, H.; Kawakita, K.; Tsuragi, H.; Mashima, K. 1,4-Bis(trimethylsilyl)-1,4-diaza-2,5-cyclohexadienes as strong salt-free reductants for generating low-valent early transition metals with electron-donating ligands. J. Am. Chem. Soc. 2014, 136, 5161–5170. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W. Effects of cyclic 8π-electron conjugation in reductively silylated nitrogen heterocycles. J. Am. Chem. Soc. 1983, 105, 707–713. [Google Scholar] [CrossRef]
- Majhi, P.K.; Ikeda, H.; Sasamori, T.; Tsuragi, H.; Mashima, K.; Tokitoh, N. Inorganic-salt-free reduction in main-group chemistry: Synthesis of a dibismuthene and a distibene. Organometallics 2017, 36, 1224–1226. [Google Scholar] [CrossRef]
- Cui, H.; Shao, Y.; Li, X.; Kong, L.; Cui, C. Dehydrochlorination to silylenes by N-heterocyclic carbenes. Organometallics 2009, 28, 5191–5195. [Google Scholar] [CrossRef]
- Jana, A.; Tavčar, G.; Roesky, H.; Schulzke, C. Facile synthesis of dichlorosilane by metathesis reaction and dehydrogenation. Dalton Trans. 2010, 39, 6217–6220. [Google Scholar] [CrossRef] [PubMed]
- Gans-Eichler, T.; Gudat, D.; Nӓttinen, K.; Nieger, M. The transfer of tin and germanium atoms from N-heterocyclic stannylenes and germylenes to diazadienes. Chem. Eur. J. 2006, 12, 1162–1173. [Google Scholar] [CrossRef] [PubMed]
- Gans-Eichler, T.; Gudat, D.; Nieger, M. Tin analogues of “arduengo carbenes”: Synthesis of 1,3,2λ2-diazastannoles and transfer of Sn atoms between a 1,3,2λ2-diazastannole and a diazadiene. Angew. Chem. Int. Ed. 2002, 41, 1888–1891. [Google Scholar] [CrossRef]
- Mansell, S.M.; Russel, C.A.; Wass, D.F. Synthesis of chelating diamido Sn(IV)compounds from oxidation of Sn(II) and directly from Sn(IV) precursors. Dalton Trans. 2015, 44, 9756–9765. [Google Scholar] [CrossRef] [PubMed]
- Sarazin, Y.; Coles, S.J.; Hughes, D.L.; Hursthouse, M.B.; Bochmann, M. Cationic brønsted acids for the preparation of SnIV salts: Synthesis and characterization of [Ph3Sn(OEt2)][H2N{B(C6F5)3}2], [Sn(NMe2)3(HNMe2)2][B(C6F5)4] and [Me3Sn(HNMe2)2][B(C6F5)4]. Eur. J. Inorg. Chem. 2006, 3211–3220. [Google Scholar] [CrossRef]
- Lichtblau, A.; Eblend, A.; Hausen, H.-D.; Kaim, W. N,N’-disilylated 1,4-dihydropyrazines: Organosilyl substitution reactions, structural effects of steric hindrance, and electron exchange with C60. Chem. Ber. 1995, 128, 745–750. [Google Scholar] [CrossRef]
- Rej, S.; Pramanik, S.; Tsurugi, H.; Mashima, K. Dehalogenation of vicinal dihalo compounds by 1,1′-bis(trimethylsilyl)-1H, 1′H-4,4′-bipyridinylidene for giving alkenes and alkynes in a salt-free manner. Chem. Commun. 2017, 53, 13157–13160. [Google Scholar] [CrossRef] [PubMed]
- Koten, G.V.; Vrieze, K. 1,4-Diaza-1,3-butadiene (α-diimine) ligands: Their coordination modes and the reactivity of their metal complexes. Adv. Organomet. Chem. 1982, 21, 151–239. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raut, R.K.; Amin, S.F.; Sahoo, P.; Kumar, V.; Majumdar, M. One-Pot Synthesis of Heavier Group 14 N-Heterocyclic Carbene Using Organosilicon Reductant. Inorganics 2018, 6, 69. https://doi.org/10.3390/inorganics6030069
Raut RK, Amin SF, Sahoo P, Kumar V, Majumdar M. One-Pot Synthesis of Heavier Group 14 N-Heterocyclic Carbene Using Organosilicon Reductant. Inorganics. 2018; 6(3):69. https://doi.org/10.3390/inorganics6030069
Chicago/Turabian StyleRaut, Ravindra K., Sheikh Farhan Amin, Padmini Sahoo, Vikas Kumar, and Moumita Majumdar. 2018. "One-Pot Synthesis of Heavier Group 14 N-Heterocyclic Carbene Using Organosilicon Reductant" Inorganics 6, no. 3: 69. https://doi.org/10.3390/inorganics6030069
APA StyleRaut, R. K., Amin, S. F., Sahoo, P., Kumar, V., & Majumdar, M. (2018). One-Pot Synthesis of Heavier Group 14 N-Heterocyclic Carbene Using Organosilicon Reductant. Inorganics, 6(3), 69. https://doi.org/10.3390/inorganics6030069