Preparation and DFT Studies of κ2C,N-Hypercoordinated Oxazoline Organotins: Monomer Constructs for Stable Polystannanes


Abstract
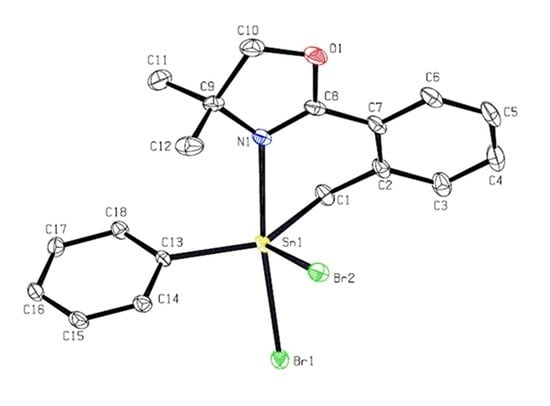
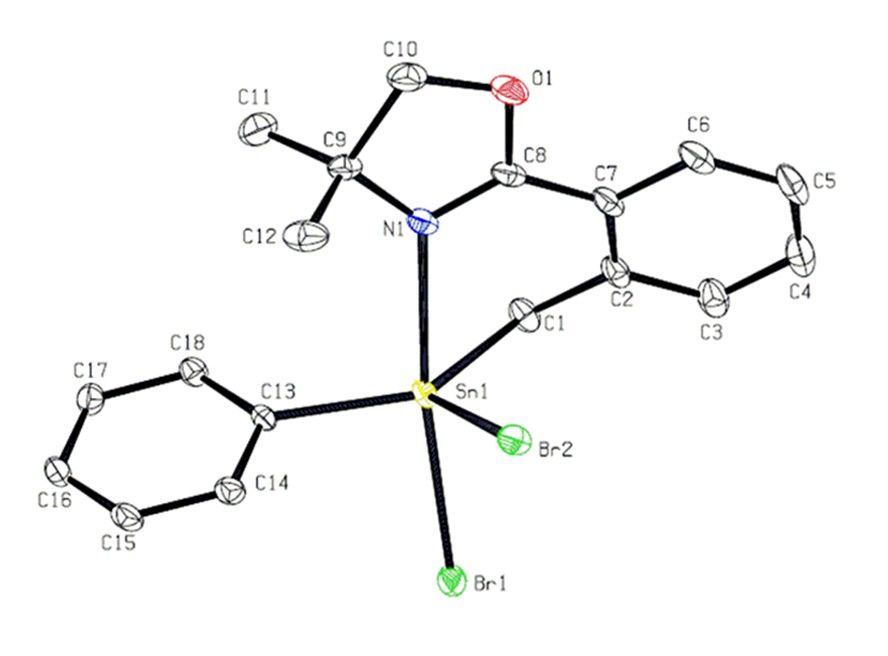
:
1. Introduction
2. Results
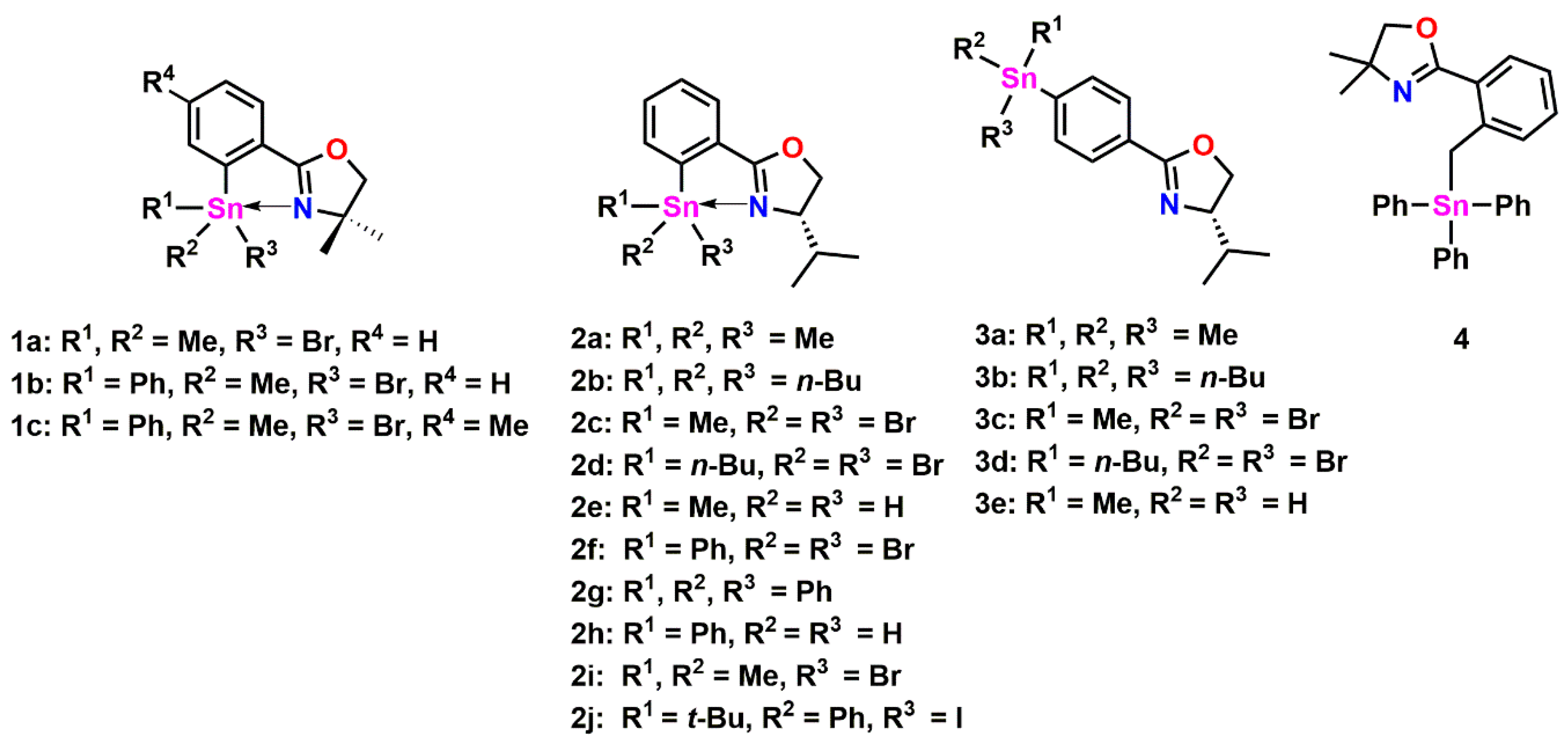
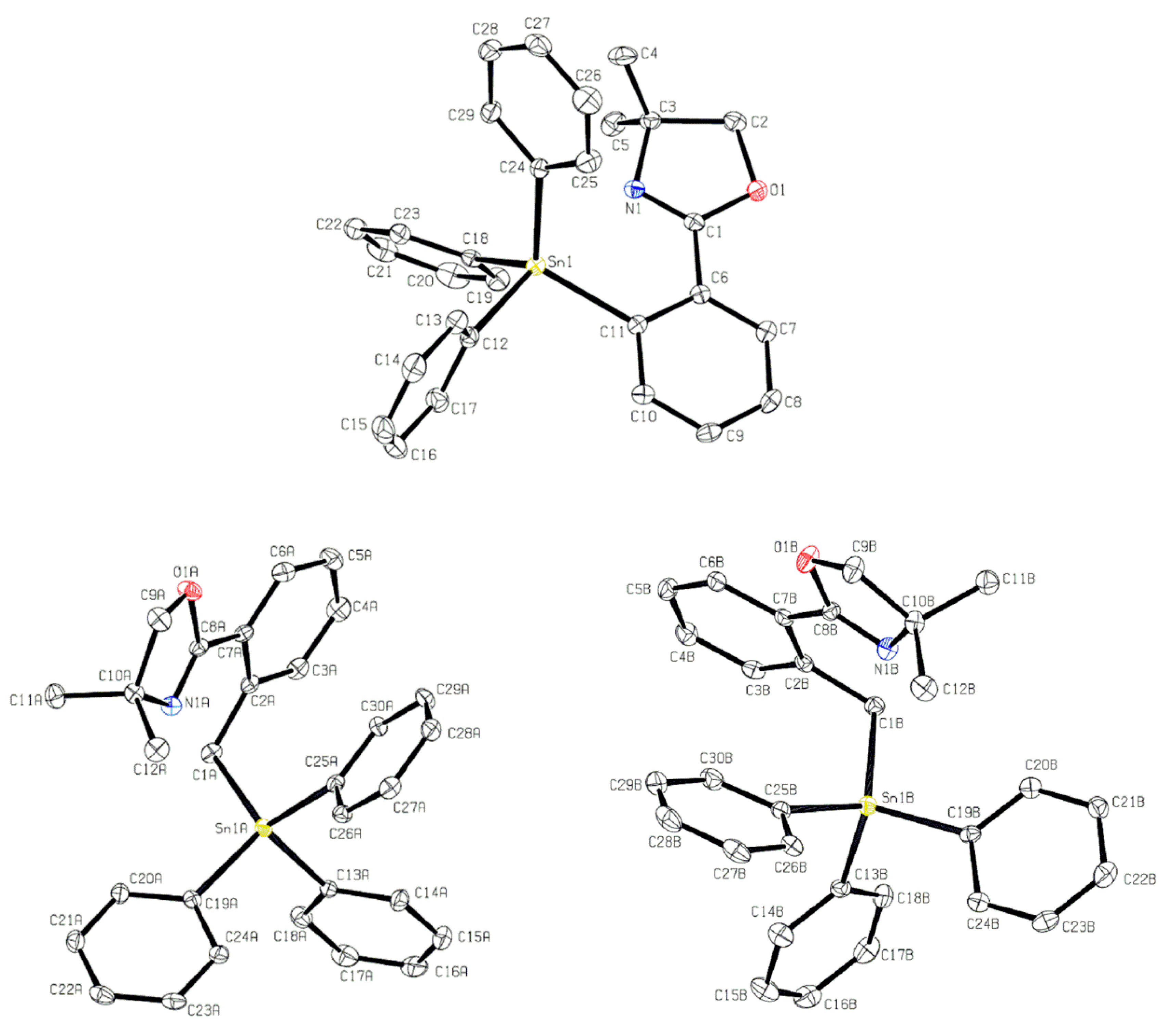
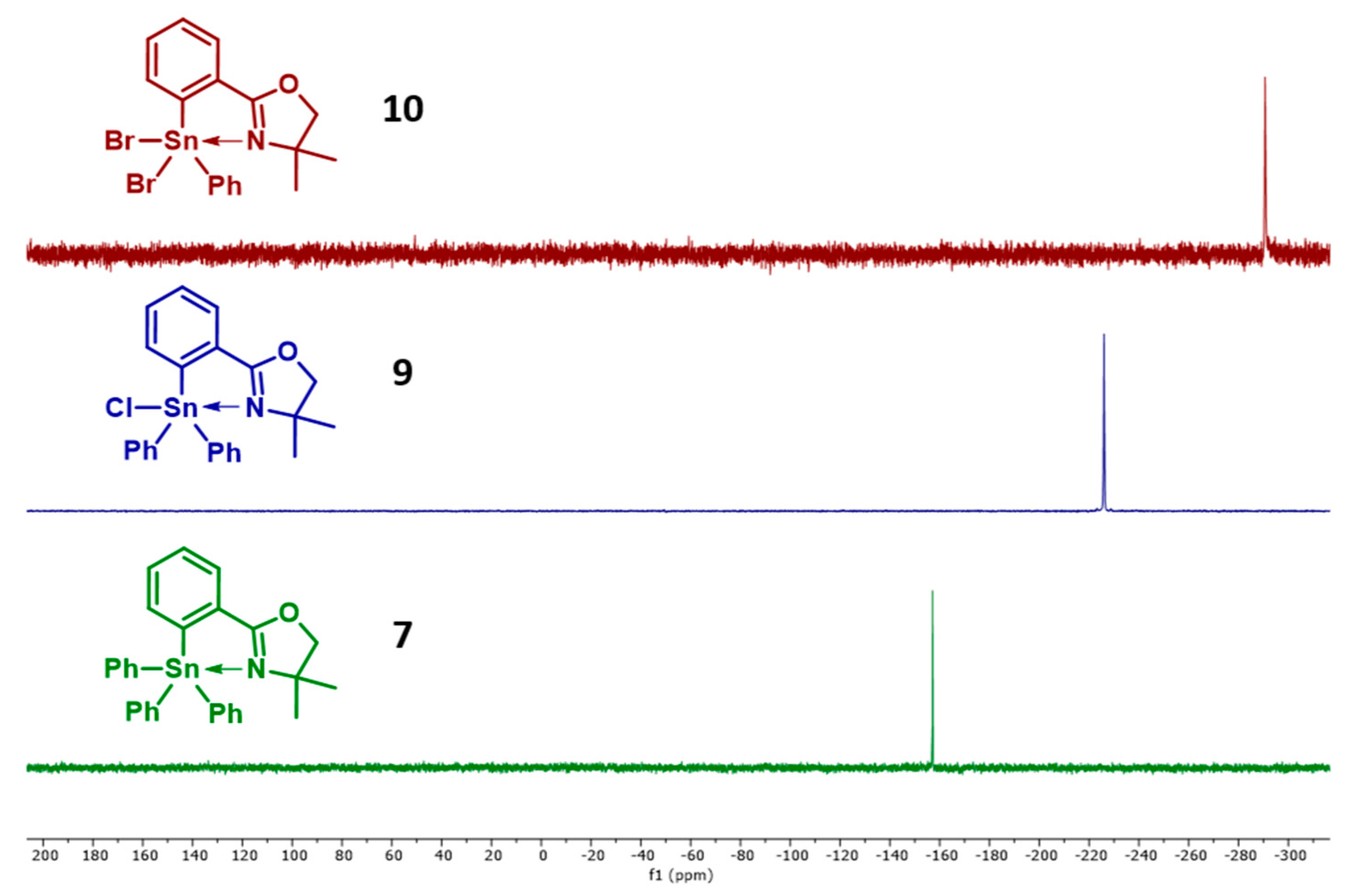
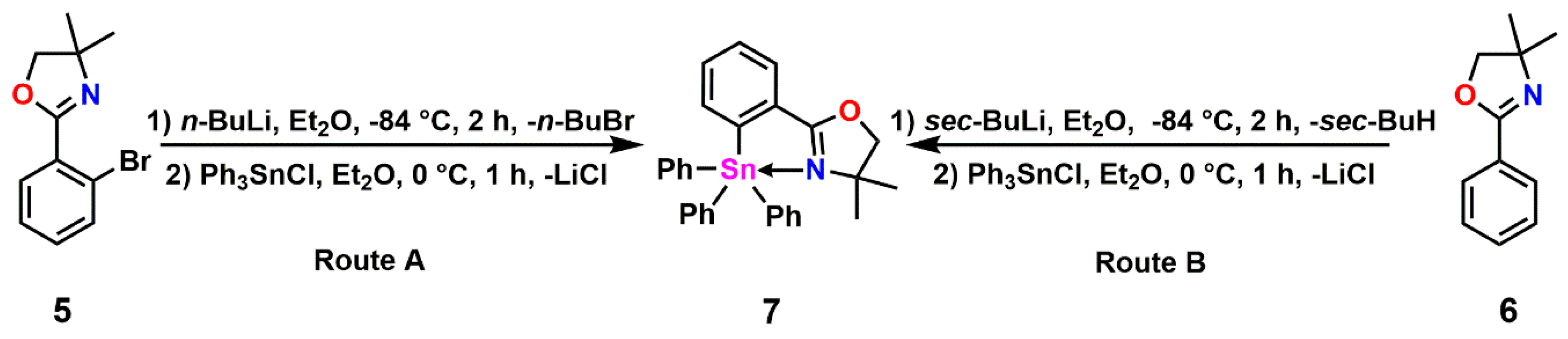
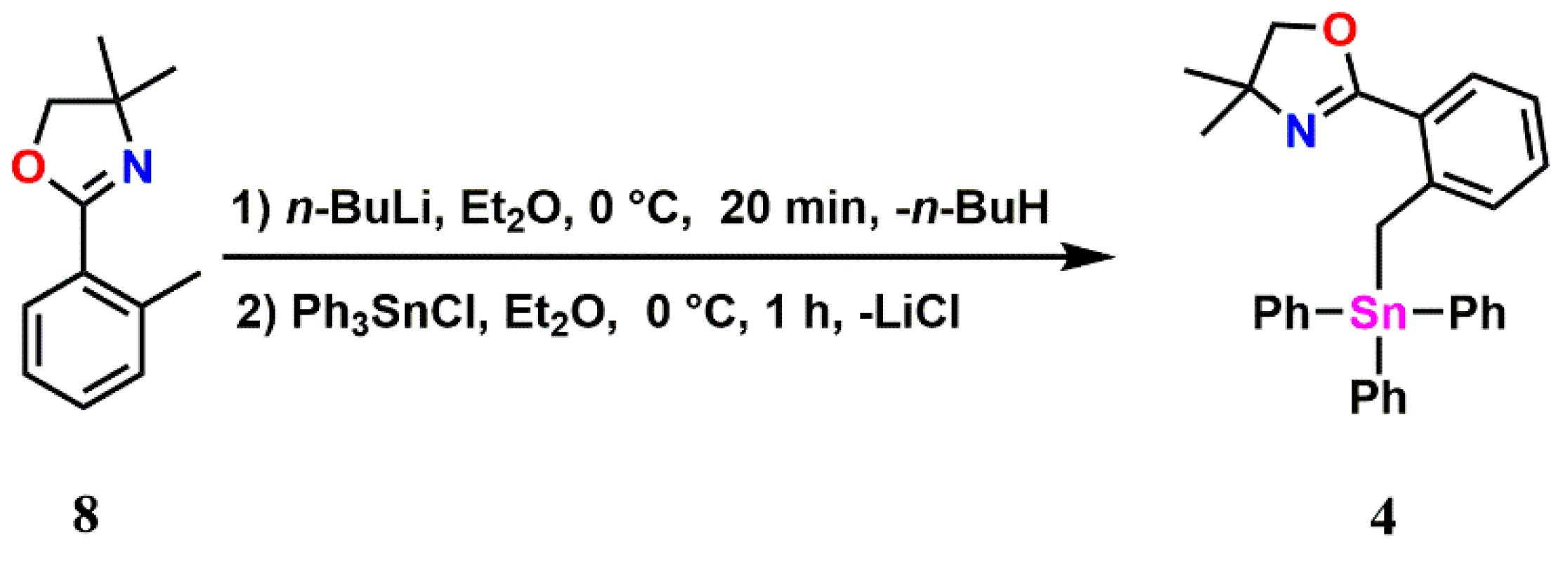
2.1. Triphenyl Oxazoline Stannanes
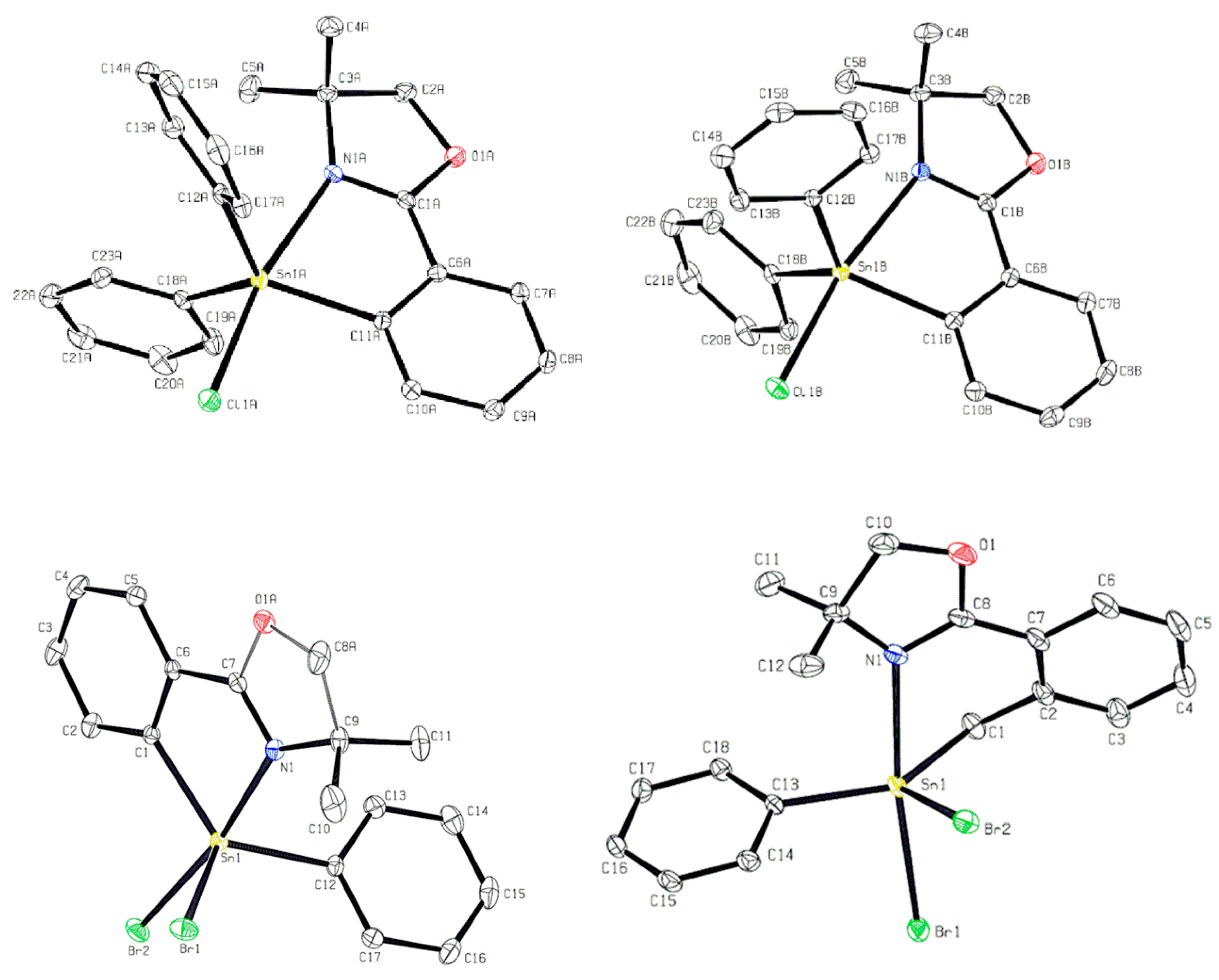
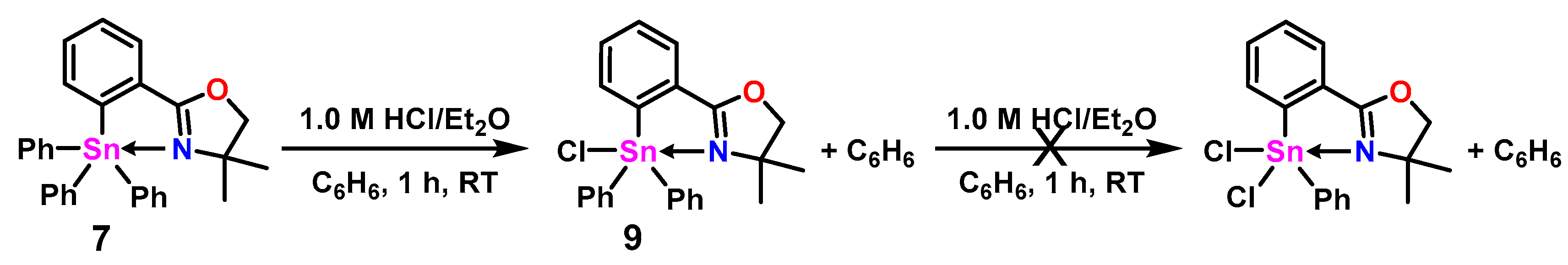
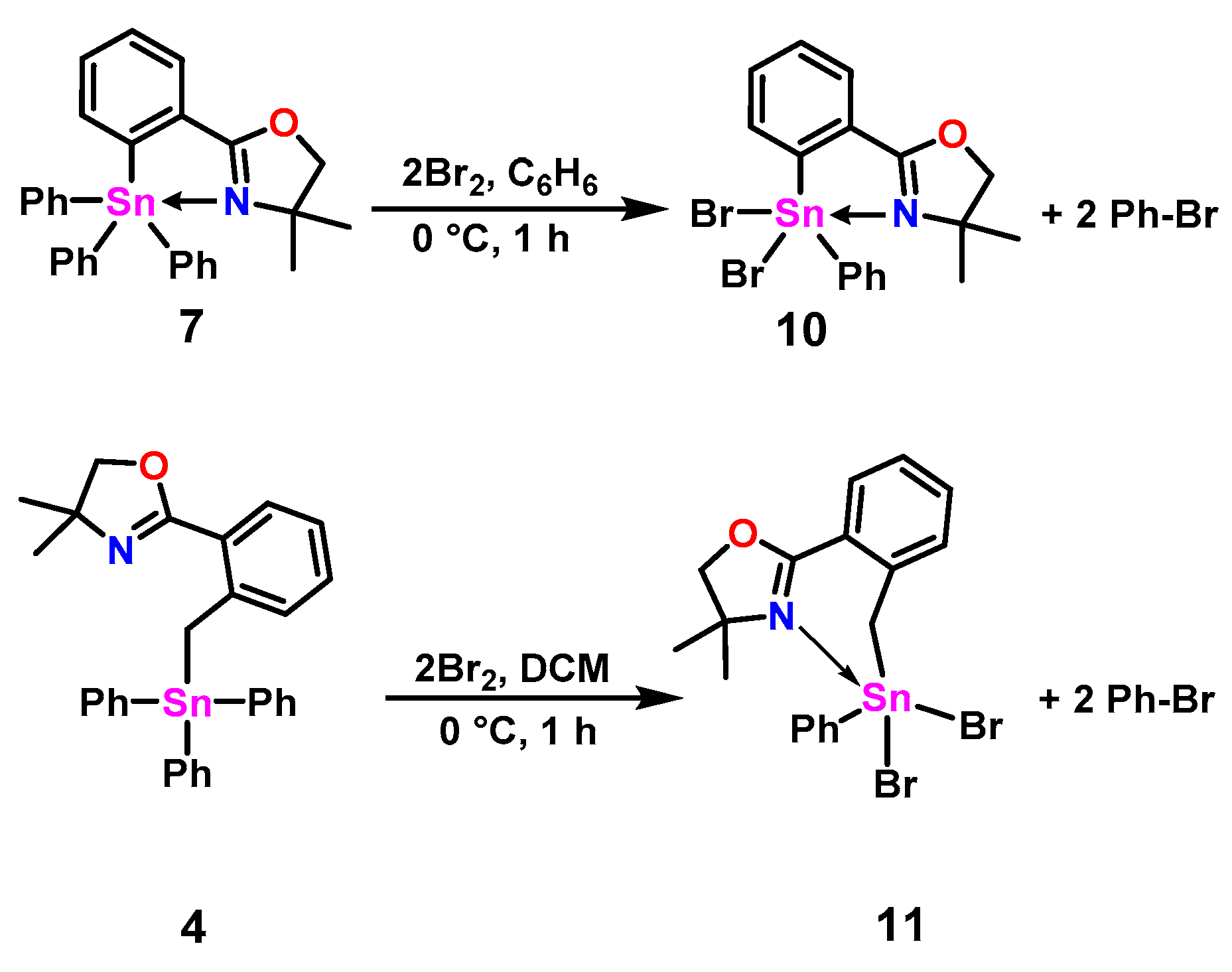
2.2. Halogenated Oxazoline Stannanes
2.3. DFT Studies
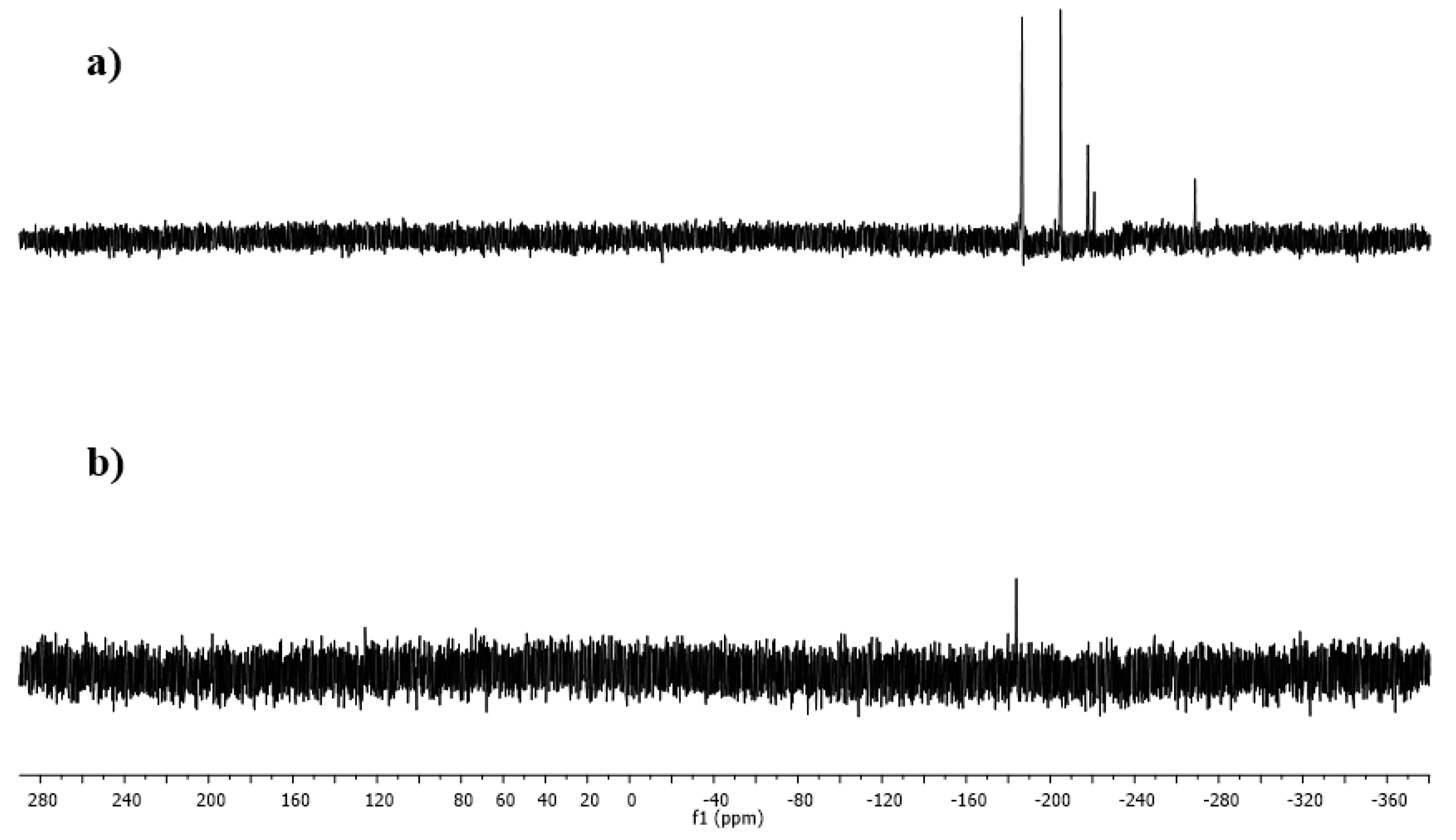
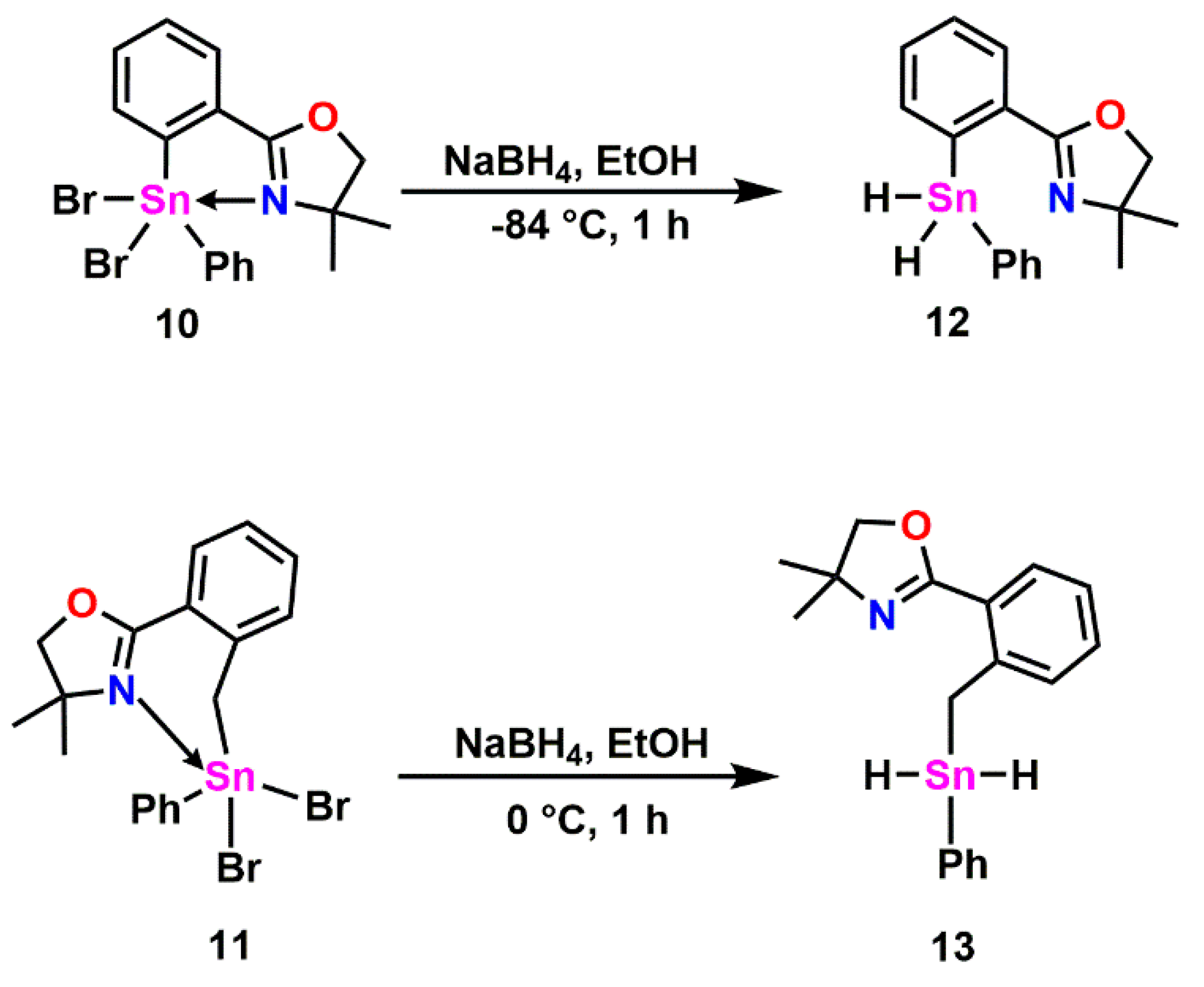
2.4. Oxazoline Stannane Dihydrides
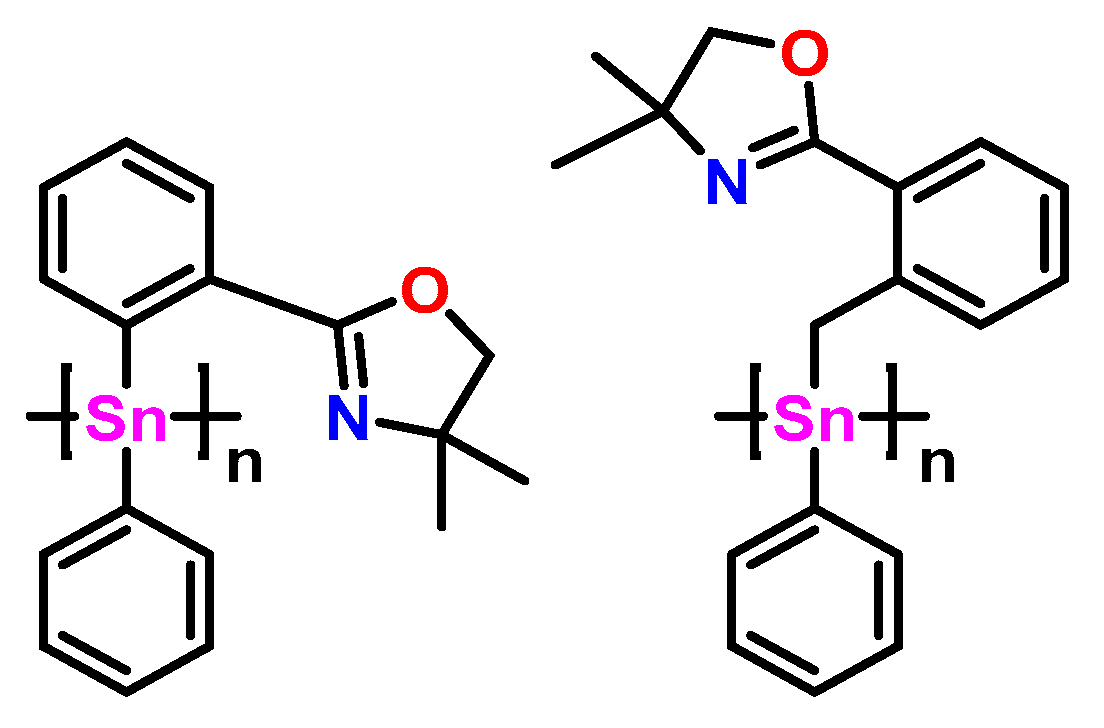
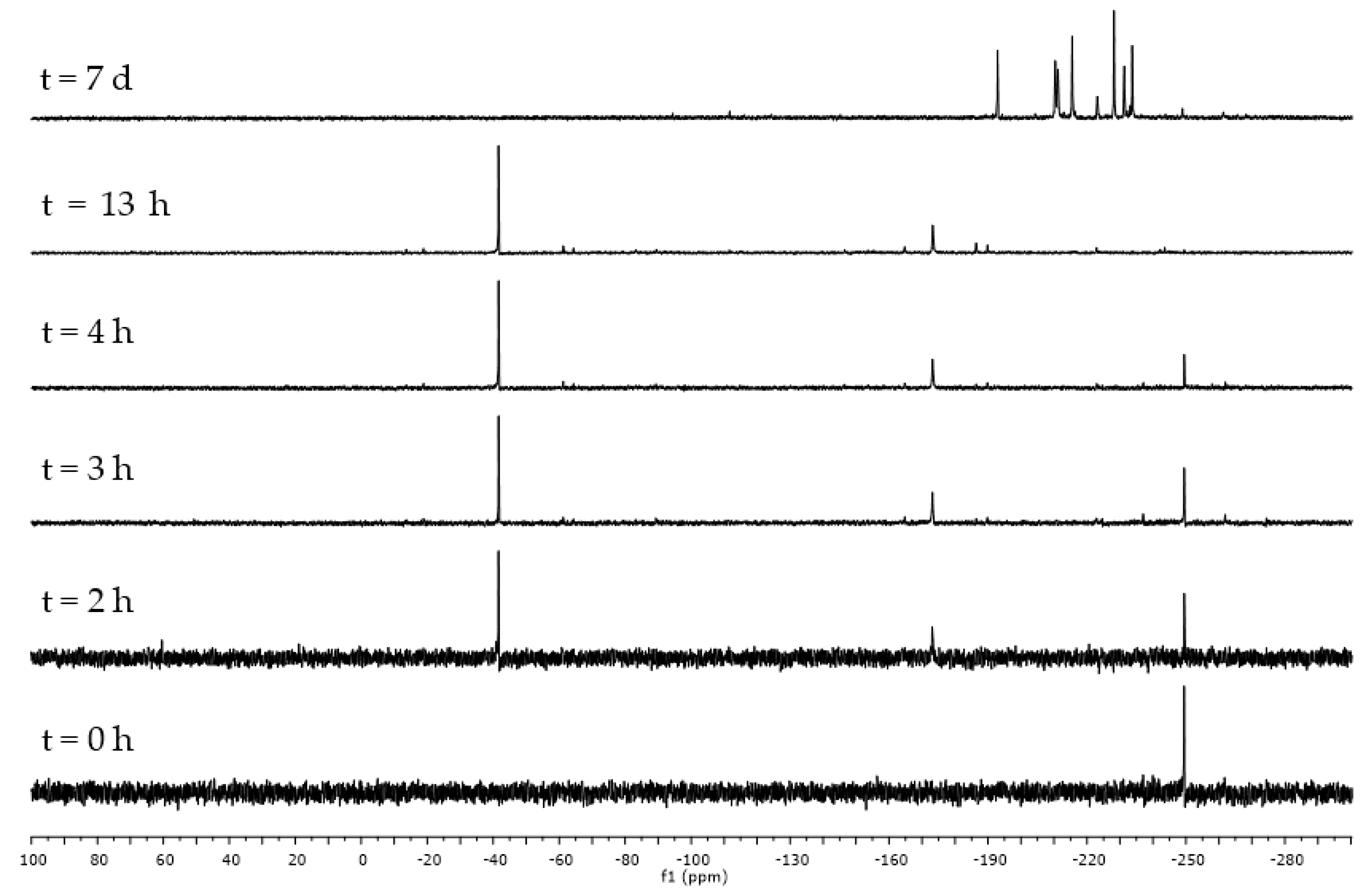
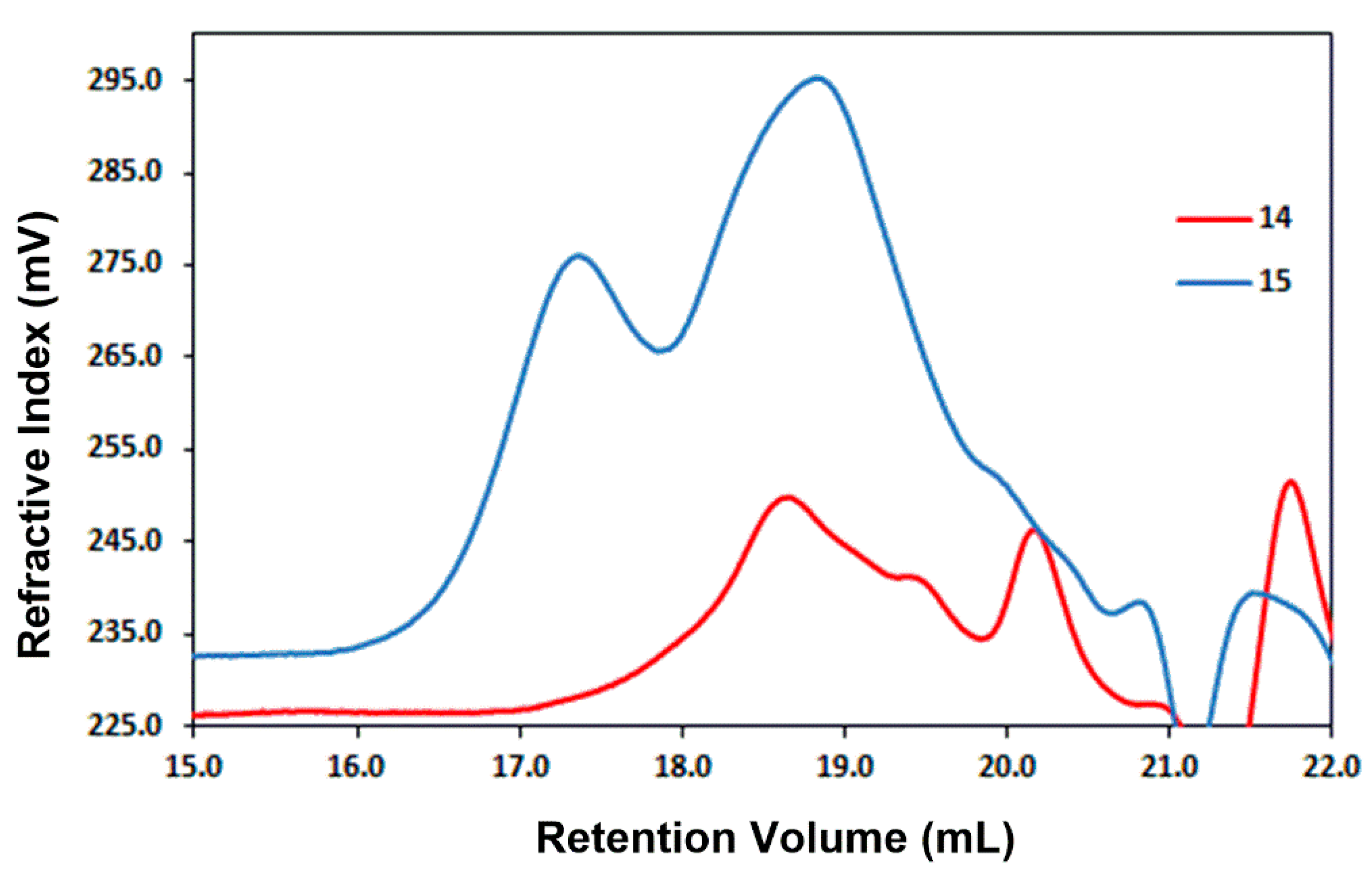
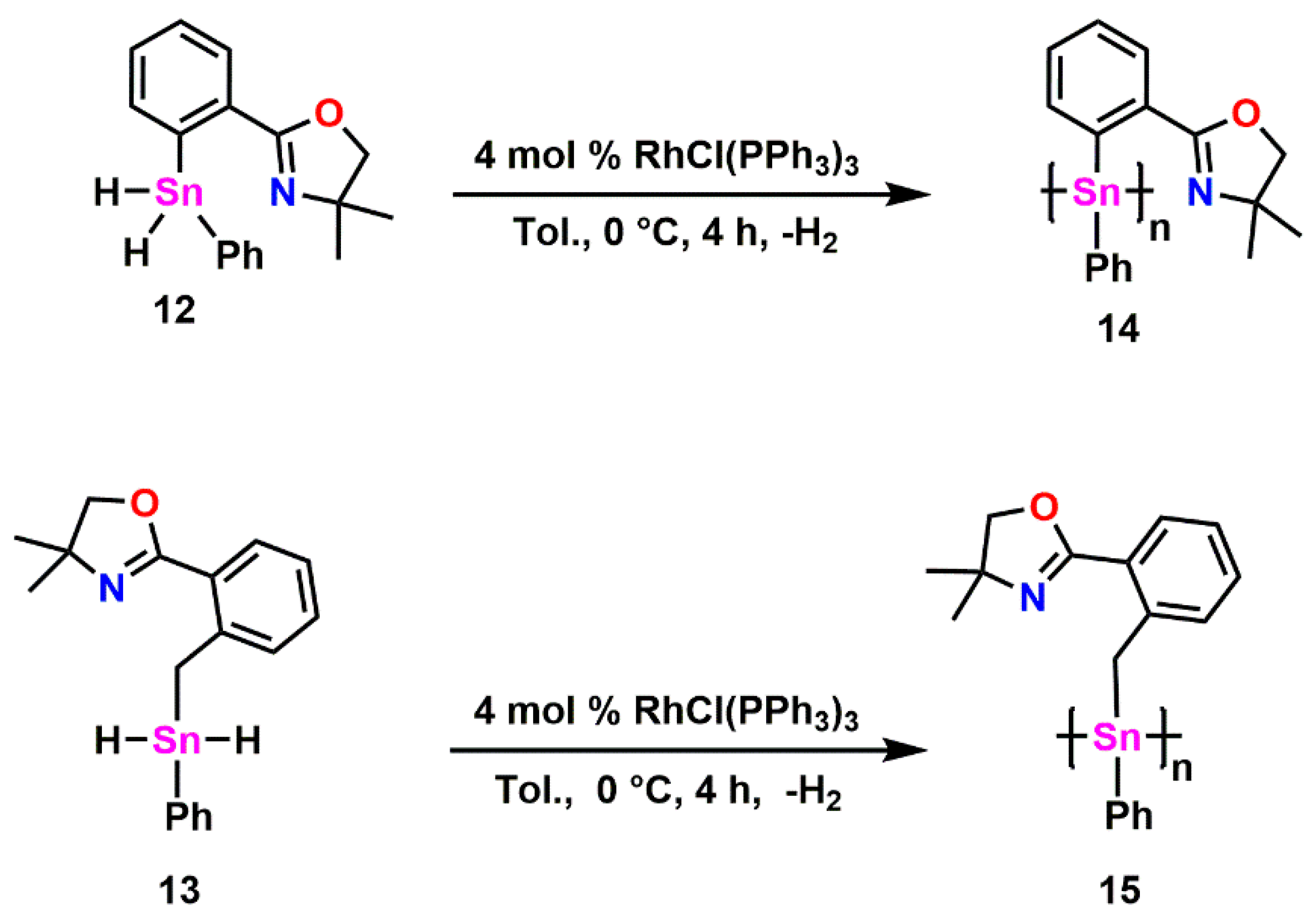
2.5. Synthesis of Polymers 14 and 15
3. Materials and Methods
3.1. General Considerations
3.2. Computational Details
3.3. Synthesis of 2-(2-Bromophenyl)-4,4-dimethyl-4,5-dihydrooxazole (5)
3.4. Synthesis of 4,4-Dimethyl-2-(2-(triphenylstannyl)phenyl)-4,5-dihydrooxazole (7):
3.5. Synthesis of 2-(2-(Chlorodiphenylstannyl)phenyl)-4,4-dimethyl-4,5-dihydrooxazole (9)
3.6. Synthesis of 2-(2-(Dibromo(phenyl)stannyl)phenyl)-4,4-dimethyl-4,5-dihydrooxazole (10)
3.7. Synthesis of 4,4-Dimethyl-2-(2-(phenylstannyl)phenyl)-4,5-dihydrooxazole (12)
3.8. Synthesis of Polymer (14)
3.9. Synthesis of 4,4-Dimethyl-2-(o-tolyl)-4,5-dihydrooxazole (8)
3.10. Synthesis of 4,4-Dimethyl-2-(2-((triphenylstannyl)methyl)phenyl)-4,5-dihydrooxazole (4)
3.11. Synthesis of 2-(2-((Dibromo(phenyl)stannyl)methyl)phenyl)-4,4-dimethyl-4,5-dihydrooxazole (11)
3.12. Synthesis of 4,4-Dimethyl-2-(2-((phenylstannyl)methyl)phenyl)-4,5-dihydrooxazole (13)
3.13. Synthesis of Polymer (15)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Caseri, W. Polystannanes: Processible molecular metals with defined chemical structures. Chem. Soc. Rev. 2016, 45, 5187–5199. [Google Scholar] [CrossRef] [PubMed]
- Deacon, P.R.; Devylder, N.; Hill, M.S.; Mahon, M.F.; Molloy, K.C.; Price, G.J. Organotin compounds bearing mesogenic sidechains: Synthesis, X-ray structures and polymerisation chemistry. J. Organomet. Chem. 2003, 687, 46–56. [Google Scholar] [CrossRef]
- Lu, V.Y.; Tilley, T.D. Poly(diaryl)stannanes: Influence of substituents on the σ–σ* transition energy. Macromolecules 2000, 33, 2403–2412. [Google Scholar] [CrossRef]
- Harrypersad, S.; Foucher, D. Alternating polystannanes: Syntheses and properties. Chem. Commun. 2015, 51, 7120–7123. [Google Scholar] [CrossRef]
- Choffat, F.; Buchmüller, Y.; Mensing, C.; Smith, P.; Caseri, W. Poly(di(ω-alkylphenyl)stannane)s. J. Inorg. Organomet. Polym. Mater. 2009, 19, 166–175. [Google Scholar] [CrossRef]
- Imori, T.; Lu, V.; Cai, H.; Tilley, T.D. Metal-Catalyzed Dehydropolymerization of Secondary Stannanes to High Molecular Weight Polystannanes. J. Am. Chem. Soc. 1995, 117, 9931–9940. [Google Scholar] [CrossRef]
- Pau, J.; Lough, A.J.; Wylie, R.S.; Gossage, R.A.; Foucher, D.A. Proof of Concept Studies Directed Towards Designed Molecular Wires: Property Driven Synthesis of Air and Moisture-Stable Polystannanes. Chem. A Eur. J. 2017, 57, 14367–14374. [Google Scholar] [CrossRef]
- Pau, J.; D’Amaral, G.M.; Lough, A.J.; Wylie, R.S.; Foucher, D.A. Synthesis and Characterization of Readily Modified Poly(aryl)(alkoxy)stannanes by use of Hypercoordinated Sn Monomers. Chem. A Eur. J. 2018, 24, 18762–18771. [Google Scholar] [CrossRef]
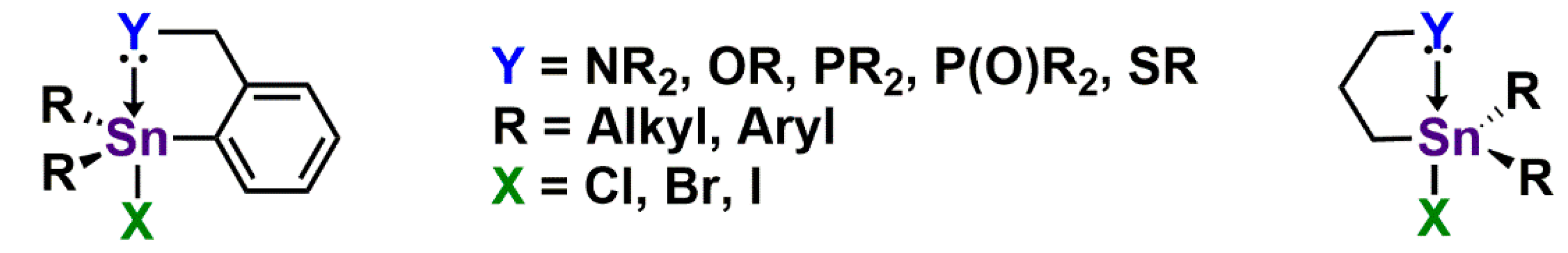
- Khan, A.; Foucher, D.A. Hypercoordinate compounds of the group 14 elements containing κ2-C,N-, C,O-, C,S- and C,P-ligands. Coord. Chem. Rev. 2016, 312, 41–66. [Google Scholar] [CrossRef]
- Musher, J.I. The Chemistry of Hypervalent Molecules. Angew. Chem. Int. Ed. Engl. 1969, 8, 54–68. [Google Scholar] [CrossRef]
- Minyaev, R.M.; Gribanova, T.N.; Minkin, V.I. Comprehensive Inorganic Chemistry II, 2nd ed.; Reedijk, J., Poeppelmeier, K., Eds.; Elsevier, Ltd.: Oxford, UK, 2013; Volume 9, pp. 109–132. [Google Scholar]
- Nandi, A.; Kozuch, S. History and Future of Dative Bonds. Chem. A Eur. J. 2020, 26, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Van Koten, G.; Noltes, J.G.; Spek, A.L. Crystal and Molecular Structure of C,N-{2-[(dimethylamino)phenyl} diphenyltin bromide J. Organomet. Chem. 1976, 118, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Boyer, J.; Breliere, C.; Corriu, R.J.P.; Kpoton, A.; Poirier, M.; Royo, G. Enhancement of Si–H bond reactivity in pentacooridinated structures. J. Organomet. Chem. 1986, 311, C39–C43. [Google Scholar] [CrossRef]
- Breliere, C.; Carre, F.; Corriu, R.J.P.; De Saxce, A.; Poirier, M.; Royo, G. Pentacoordinate silicon and germanium derivatives: Molecular structure. J. Organomet. Chem. 1981, 205, C1–C3. [Google Scholar] [CrossRef]
- De Wit, P.P.; Van Der Kooi, H.O.; Wolters, J. An Intriguing plumbylene: Bis[2-(dimethylaminomethyl)-phenyl]lead. J. Organomet. Chem. 1981, 216, 9–11. [Google Scholar] [CrossRef]
- Khan, A.; Pau, J.; Loungxay, J.; Magobenny, T.; Wylie, R.S.; Lough, A.J.; Foucher, D. Hypercoordinated organotin(IV) compounds containing C,O- and C,N- chelating ligands: Synthesis, characterisation, DFT studies and polymerization behavior. J. Organomet. Chem. 2019, 900, 120910. [Google Scholar] [CrossRef]
- Meyers, A.I.; Mihelich, E.D. The Synthetic Utility of 2-Oxazolines. Angew. Chem. Int. Ed. 1976, 15, 270–281. [Google Scholar] [CrossRef]
- Gomez, M.; Muller, G.; Rocamora, M. Coordination chemistry of oxazoline ligands. Coord. Chem. Rev. 1999, 193–195, 769–835. [Google Scholar] [CrossRef]
- Adjei, J.A.; Lough, A.J.; Gossage, R.A. Synthesis and characterisation of C,N κ2-N,O oxazoline-enolate complexes of nickel(II): Explorations in coordination chemistry and metal mediated polymerization. RSC Adv. 2019, 9, 3956–3964. [Google Scholar] [CrossRef] [Green Version]
- Adams, N.; Schubert, U.S. Poly(2-oxazolines) in biological and biomedical application contexts Adv. Drug Deliv. Rev. 2007, 59, 1504–1520. [Google Scholar] [CrossRef]
- Krolikiewicz, H.; Vorbrüggen, K. A simple synthesis of Δ2-oxazolines, Δ2-oxazines, Δ2-thiazolines and 2-substituted benzoxazoles. Tetrahedron 1993, 49, 9353–9372. [Google Scholar] [CrossRef]
- Lee, J.D.; Kim, H.S.; Han, W.S.; Kang, S.O. Chiral organotin complexes stabilized by C,N-chelating oxazolinyl-o-carboranes. J. Organomet. Chem. 2010, 695, 463–468. [Google Scholar] [CrossRef]
- Selvaratnam, S.; Mun, K.; Das, V.G.K. Synthesis and structural characterization of (2-oxazolinylthienyl) tetraorganotin (IV) compounds. J. Organomet. Chem. 1994, 464, 143–148. [Google Scholar] [CrossRef]
- Stol, M.; Snelders, D.J.M.; De Pater, J.J.M.; Van Klink, G.P.M.; Kooijman, H.; Spek, A.L.; Van Koten, G. Synthesis and structural characterization of lithium and trimethyltin complexes of 2,6-bis(oxazolinyl)phenyl. Organometallics 2005, 24, 743–749. [Google Scholar] [CrossRef] [Green Version]
- Jastrzebski, J.T.B.H.; Wehman, E.; Boersma, J.; van Koten, G. Synthesis and structure of [2-(4,4-dimethyl-2-oxazoline)-5-methylphenyl]methylphenyltin bromide. A novel triorganotin halide having a configurationally stable chiral tin center. J. Organomet. Chem. 1991, 409, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Cmoch, P.; Urbańczyk-Lipkowska, Z.; Petrosyan, A.; Stȩpień, A.; Staliński, K. The 1H, 13C, 15N and117Sn NMR study of the intramolecular Sn–N interaction in tri- and tetraorganotin compounds containing the chiral 2-(4-isopropyl-2-oxazolinyl)-5-phenyl ligand. J. Mol. Struct. 2005, 733, 29–39. [Google Scholar] [CrossRef]
- Staliński, K.; Urbańczyk-Lipkowska, Z.; Cmoch, P.; Rupnicki, L.; Grachev, A. New chiral tin compounds containing the 2-(4-isopropyl-2-oxazolinyl)-5-phenyl ligand. J. Organomet. Chem. 2006, 691, 2394–2402. [Google Scholar] [CrossRef]
- Rupnicki, L.; Urbańczyk-Lipkowska, Z.; Stȩpień, A.; Cmoch, P.; Pianowski, Z.; Staliński, K. New distannanes containing the chiral 2-(4-isopropyl-2-oxazolinyl)-5-phenyl ligand. J. Organomet. Chem. 2005, 690, 3690–3696. [Google Scholar] [CrossRef]
- Matkowska, D.; Gola, M.; Śniezek, M.; Cmoch, P.; Staliński, K. Structural assignment of organotin hydrides containing the oxazoline ligand. J. Organomet. Chem. 2007, 692, 2036–2045. [Google Scholar] [CrossRef]
- Bonnardel, P.-A.; Parish, R.V. Organomercury(II) and organotin(IV) compounds with nitrogen-containing substituents. J. Organomet. Chem 1996, 515, 221–232. [Google Scholar] [CrossRef]
- Gschwend, H.W.; Hamdan, A. Ortho-Lithiation of Aryloxazolines. J. Org. Chem. 1975, 40, 2008–2009. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, Structure, and Spectroscopic Properties of Copper(II) Compounds containing Nitrogen–Sulphur Donor Ligands; the Crystal and Molecular Structure of Aqua[l,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) Perchlorate. J. Chem. Soc. Dalton. Trans. 1984, 1349–1356. [Google Scholar] [CrossRef]
- Jurkschat, K.; Tzschach, A.; Meunier-Piret, J. Crystal and Molecular Structure of 1-Aza-5-stannan-5-methyltricyclo[3,3,3]undecane. Evidence for a transannular donor–acceptor interaction in a tetraorganotin compound. J. Organomet. Chem. 1986, 315, 45–49. [Google Scholar] [CrossRef]
- Baukov, Y.I.; Tandura, S.N.; Rappoport, Z. The Chemistry of Organic Germanium, Tin and Lead Compounds; John Wiley & Sons, Ltd.: New York, NY, USA, 2002. [Google Scholar] [CrossRef]
- Munguia, T.; Lo, M.; Cervantes-Lee, F.; Pannell, K.H. Intramolecular Chalcogen−Tin Interactions in (o-MeE-C6H4)CH2SnPh3−nCln (E = S, O; n = 0, 1, 2), Characterized by X-ray Diffraction and 119Sn Solution and Solid-State NMR. Inorg. Chem. 2007, 46, 1305–1314. [Google Scholar] [CrossRef]
- Novák, P.; Padělková, Z.; Císařová, I.; Kolářová, L.; Růžička, A.; Holeček, J. Structural study of C,N-chelated monoorganotin(IV) halides. Appl. Organomet. Chem. 2006, 20, 226–232. [Google Scholar] [CrossRef]
- Švec, P.; Růžičková, Z.; Vlasák, P.; Turek, J.F.; De Proft, F.; Růžička, A. Expanding the family of C,N-chelated organotin(IV) pseudohalides: Synthesis and structural Characterization. J. Organomet. Chem. 2016, 801, 14–23. [Google Scholar] [CrossRef]
- Whittleton, S.R.; Boyd, R.J.; Grindley, T.B. Evaluation of effective core potentials and basis sets for the prediction of the geometries of alkyltin halides. J. Phys. Chem. A 2006, 110, 5893–5896. [Google Scholar] [CrossRef]
- Matczak, P. Assessment of B3LYP combined with various ECP basis sets for systems containing Pd, Sn, and Pb. Comput. Theor. Chem. 2012, 983, 25–30. [Google Scholar] [CrossRef]
- Perdew, J.P. Unified theory of exchange and correlation beyond the local density approximation. In Electronic Structure of Solids’ 91; Ziesche, P., Eschig, H., Eds.; Akademie Verlag: Berlin, Germany, 1991; pp. 11–20. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Goerigk, L.; Mehta, N. A Trip to the Density Functional Theory Zoo: Warnings and Recommendations for the User. Aust. J. Chem. 2019, 72, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef] [PubMed]
- Goerigk, L.; Grimme, S. A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions. Phys. Chem. Chem. Phys. 2011, 13, 6670–6688. [Google Scholar] [CrossRef] [PubMed]
- Choffat, F.; Smith, P.; Caseri, W. Polystannanes: Polymers of a Molecular, Jacketed Metal-Wire Structure. Adv. Mater. 2008, 20, 2225–2229. [Google Scholar] [CrossRef]
- Khan, A.; Patel, A.; Komejan, S.; Lombardi, C.; Lough, A.J.; Foucher, D.A. Reduction of C,O-chelated organotin(IV) dichlorides and dihydrides leading to protected polystannanes. J. Organomet. Chem. 2015, 776, 180–191. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision C.01; Gaussian. Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Schöler, S.; Wahl, M.H.; Wurster, N.I.C.; Puls, A.; Hättig, C.; Dyker, G. Bidentate cycloimidate palladium complexes with aliphatic and aromatic anagostic bonds. Chem. Commun. 2014, 50, 5909–5911. [Google Scholar] [CrossRef]
- Sedelmeier, J.; Hammerer, T.; Bolm, C. C1-Symmetric Oxazolinyl Sulfoximines as Ligands in Copper-Catalyzed Asymmetric Mukaiyama Aldol Reactions. Org. Lett. 2008, 10, 917–920. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Lengths of 7 (Å) | Bond Lengths of 4′ (Å) | Bond Lengths of 4″ (Å) | |||
| Sn1–N1 | 2.762(1) | Sn1A–N1A | 3.176(4) | Sn1B–N1B | 3.234(4) |
| Sn1–C11 | 2.1602(16) | Sn1A–C1A | 2.162(4) | Sn1B–C1B | 2.153(4) |
| Sn1–C12 | 2.1752(17) | Sn1A–C13A | 2.158(4) | Sn1B–C13B | 2.155(4) |
| Sn1–C18 | 2.1378(16) | Sn1–C19A | 2.150(4) | Sn1B–C19B | 2.139(4) |
| Sn1–C24 | 2.1425(17) | Sn1–C25A | 2.143(4) | Sn1–C25B | 2.144(4) |
| Bond Angles of 7 (°) | Bond Angles of 4′ (°) | Bond Angles of 4″ (°) | |||
| N1–Sn1–C11 | 69.83(6) | C1A–Sn1A–C13A | 103.61(16) | C1B–Sn1B–C13B | 103.30(16) |
| N1–Sn1–C12 | 172.20(6) | C1A–Sn1A–C19A | 113.46(17) | C1B–Sn1B–C19B | 112.07(16) |
| N1–Sn1–C18 | 82.20(6) | C1A–Sn1A–C25A | 115.46(16) | C1B–Sn1B–C25B | 116.09(16) |
| N1–Sn1–C24 | 81.81(6) | C13A–Sn1A–C19A | 108.28(16) | C13B–Sn1B–C19B | 108.41(17) |
| C11–Sn1–C18 | 117.09(6) | C13A–Sn1A–C25A | 104.86(16) | C13B–Sn1B–C25B | 105.81(17) |
| C12–Sn1–C18 | 102.03(6) | C19A–Sn1A–C25A | 110.34(15) | C19B–Sn1B–C25B | 110.34(15) |
| Bond Lengths of 9′ (Å) | Bond Lengths of 9″ (Å) | ||
| Sn1A–N1A | 2.4658(14) | Sn1B–N1B | 2.4502(14) |
| Sn1A–C11A | 2.1401(16) | Sn1B–C11B | 2.1422(17) |
| Sn1A–C12A | 2.1322(16) | Sn1B–C12B | 2.1330(17) |
| Sn1–C18A | 2.1250 (17) | Sn1B–C18B | 2.1232(17) |
| Sn1–Cl1A | 2.4832(5) | Sn1–Cl1B | 2.4955(5) |
| Bond Angles of 9′ (°) | Bond Angles of 9″ (°) | ||
| N1A–Sn1A–Cl1A | 169.33(3) | N1B–Sn1B–Cl1B | 170.40(4) |
| N1A–Sn1A–C11A | 75.01(6) | N1B–Sn1B–C11B | 75.24(6) |
| N1A–Sn1A–C12A | 91.72(6) | N1B–Sn1B–C12B | 91.89(6) |
| N1A–Sn1A–C18A | 90.31(6) | N1B–Sn1B–C18B | 89.02(6) |
| C11A–Sn1A–ClA | 94.36(5) | C11B–Sn1B–ClB | 95.55(5) |
| C12A–Sn1A–ClA | 94.70(5) | C12B–Sn1B–ClB | 94.97(5) |
| Bond Lengths of 10 (Å) | Bond Lengths of 11 (Å) | ||
| Sn1–N1 | 2.383(3) | Sn1–N1 | 2.4245(18) |
| Sn1–C1 | 2.137(2) | Sn1–C1 | 2.127(2) |
| Sn1–C12 | 2.127(2) | Sn1–C13 | 2.1302(19) |
| Sn1–Br1 | 2.4943(3) | Sn1–Br1 | 2.5167(3) |
| Sn1–Br2 | 2.6180(3) | Sn1–Br2 | 2.6394(3) |
| Bond Angles of 10 (°) | Bond Angles of 11 (°) | ||
| N1–Sn1–Br2 | 171.36(5) | N1–Sn1–Br2 | 84.68(4) |
| N1–Sn1–Br1 | 87.98(5) | N1–Sn1–Br1 | 172.89(4) |
| N1–Sn1–C1 | 76.06(6) | N1–Sn1–C1 | 78.24(8) |
| N1–Sn1–C12 | 89.86(8) | N1–Sn1–C13 | 95.47(8) |
| C12–Sn1–C1 | 126.73(9) | C1–Sn1–C13 | 136.53(8) |
| Br1–Sn1–Br2 | 91.994(11) | Br1–Sn1–Br2 | 91.148(9) |
| Compound | B3PW91-GD3BJ | PBE0-GD3BJ | M05-2X-GD3 |
|---|---|---|---|
| 4′a,b | 0.204 | 0.186 | 0.188 |
| 4″a,b | 0.223 | 0.200 | 0.198 |
| 7c | 0.0104 | 0.00757 | 0.0120 |
| 9′a,d | 0.0229 | 0.0208 | 0.0206 |
| 9″a,d | 0.0332 | 0.0331 | 0.0335 |
| 10e | 0.0191 | 0.0141 | 0.0151 |
| 11 | 0.0526 | 0.0572 | 0.0792 |
| Complexes | 4 | 7 | 9 | 10 | 11 |
|---|---|---|---|---|---|
| CCDC number | 1987221 | 1987224 | 1987220 | 1987222 | 1987223 |
| Empirical Formula | C30H29NOSn | C29H27NOSn | C23H22ClNOSn | C17H17Br2NOSn | C18H19Br2NOSn |
| Formula Weight | 538.23 | 524.20 | 482.55 | 529.82 | 543.85 |
| Crystal System | Orthorhombic | Monoclinic | Triclinic | Monoclinic | Triclinic |
| Space Group | P212121 | P21/n | P−1 | P21/n | P−1 |
| α/Å | 8.4502(2) | 9.3351(5) | 9.1303(4) | 10.5295(7) | 8.4530(5) |
| b/Å | 18.9557(5) | 18.4709(9) | 9.3020(4) | 10.0336(5) | 10.2368(5) |
| c/Å | 31.5231(9) | 14.3598(7) | 24.2461(11) | 17.1396(11) | 11.7987(7) |
| α/° | 90 | 90 | 90.942(1) | 90 | 93.059(2) |
| β/° | 90 | 93.8460(10) | 91.775(1) | 96.667(2) | 101.936(2) |
| γ/° | 90 | 90 | 91.724(1) | 90 | 108.325(2) |
| V/Å3 | 5049.4(2) | 2470.5(2) | 2056.99(16) | 1798.50(19) | 940.50(9) |
| Z | 8 | 8 | 4 | 4 | 2 |
| Dcalc/g cm−3 | 1.416 | 1.409 | 1.558 | 1.957 | 1.920 |
| μ (Mo Kα)/Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Temperature/°K | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) |
| θ range, ° | 1.680–27.489 | 1.799–27.517 | 0.840–27.599 | 2.164–27.535 | 1.779–27.500 |
| Reflections collected | 27514 | 39843 | 66519 | 38637 | 43074 |
| Unique reflections | 11499 | 5684 | 9482 | 4135 | 4285 |
| Rint | 0.0346 | 0.0229 | 0.0237 | 0.0237 | 0.0304 |
| Residuals: R1 (I > 2 σ (I)) | 0.0316 | 0.0212 | 0.0205 | 0.0219 | 0.0183 |
| Residuals: R (All reflections) | 0.0449 | 0.0281 | 0.0255 | 0.0358 | 0.0250 |
| Residuals: wR2 (All reflections) | 0.0517 | 0.0452 | 0.0413 | 0.0422 | 0.0387 |
| Goodness of fit indicator | 0.987 | 1.105 | 1.096 | 1.052 | 1.115 |
| Max/min peak,/e Å−3 | 0.473/−0.414 | 0.542/−0.459 | 0.429/−0.600 | 0.553/−0.467 | 0.542/−494 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bender, D.N.; Lough, A.J.; Wylie, R.S.; Gossage, R.A.; Foucher, D.A. Preparation and DFT Studies of κ2C,N-Hypercoordinated Oxazoline Organotins: Monomer Constructs for Stable Polystannanes. Inorganics 2020, 8, 35. https://doi.org/10.3390/inorganics8050035
Bender DN, Lough AJ, Wylie RS, Gossage RA, Foucher DA. Preparation and DFT Studies of κ2C,N-Hypercoordinated Oxazoline Organotins: Monomer Constructs for Stable Polystannanes. Inorganics. 2020; 8(5):35. https://doi.org/10.3390/inorganics8050035
Chicago/Turabian StyleBender, Desiree N., Alan J. Lough, R. Stephen Wylie, Robert A. Gossage, and Daniel A. Foucher. 2020. "Preparation and DFT Studies of κ2C,N-Hypercoordinated Oxazoline Organotins: Monomer Constructs for Stable Polystannanes" Inorganics 8, no. 5: 35. https://doi.org/10.3390/inorganics8050035
APA StyleBender, D. N., Lough, A. J., Wylie, R. S., Gossage, R. A., & Foucher, D. A. (2020). Preparation and DFT Studies of κ2C,N-Hypercoordinated Oxazoline Organotins: Monomer Constructs for Stable Polystannanes. Inorganics, 8(5), 35. https://doi.org/10.3390/inorganics8050035