



Plants 2024, 13(20), 2883; https://doi.org/10.3390/plants13202883 - 15 Oct 2024
Viewed by 752
Abstract
►
Show Figures
Rapid Alkalization Factor (RALF) is a signaling molecule in plants that plays a crucial role in growth and development, reproductive processes, and responses to both biotic and abiotic stresses. Although RALF peptides have been characterized in Arabidopsis and rice, a comprehensive bioinformatics analysis
[...] Read more.
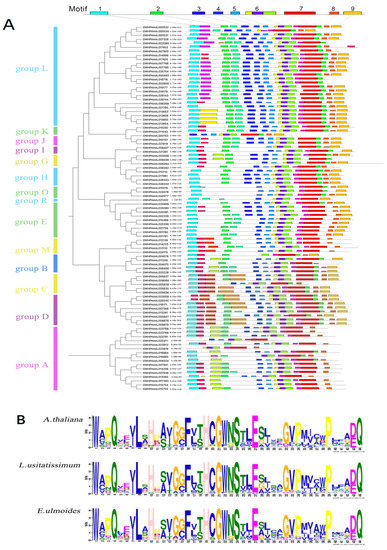
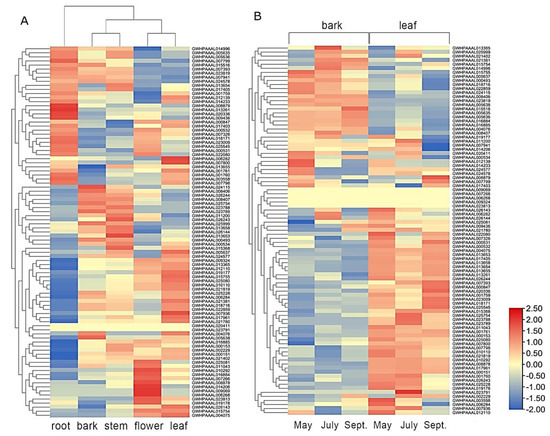
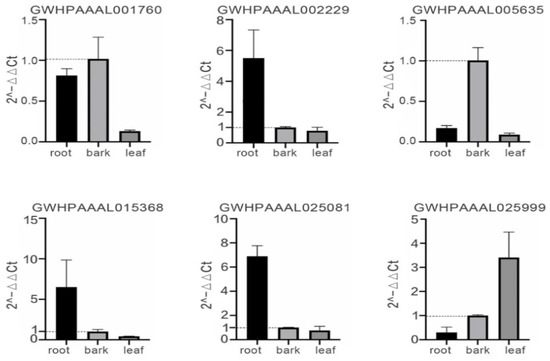
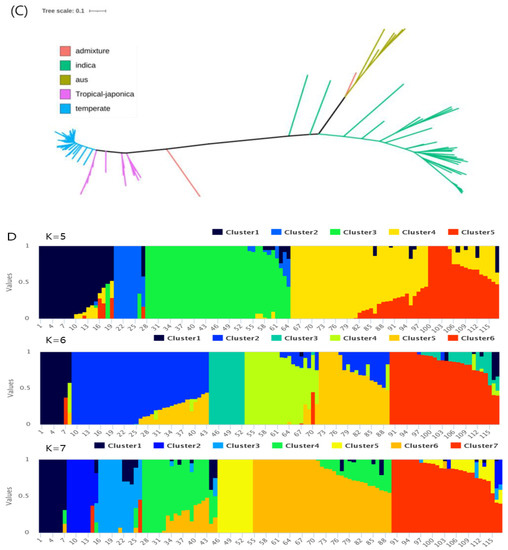
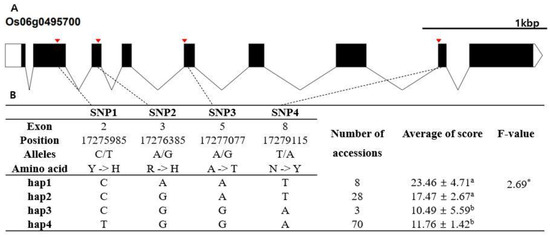
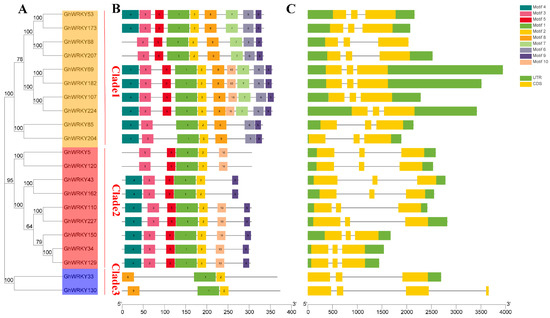
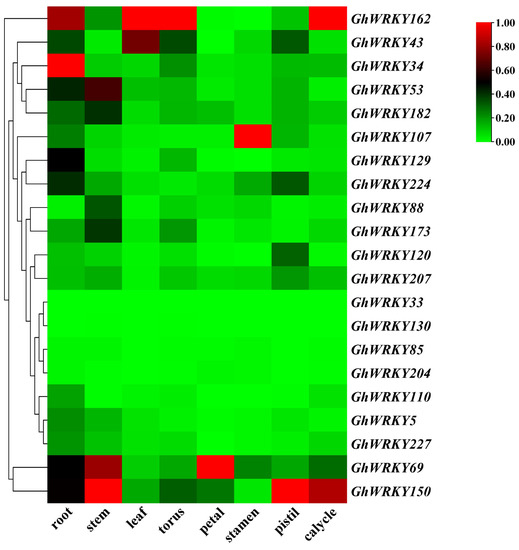
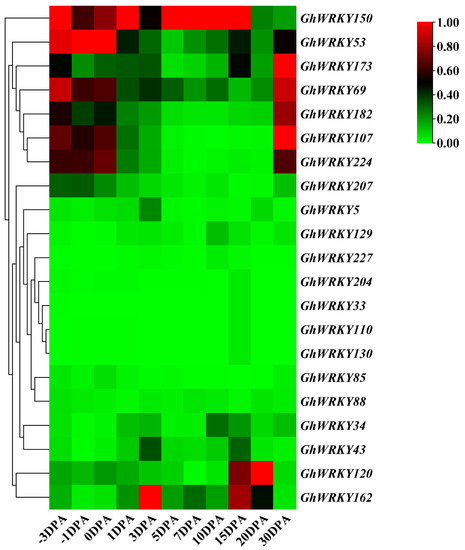
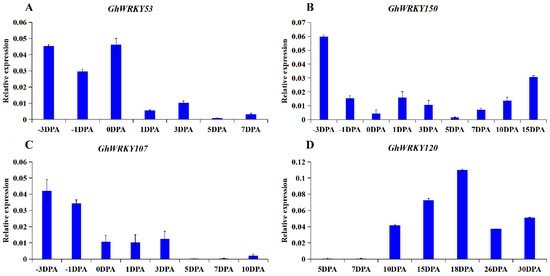
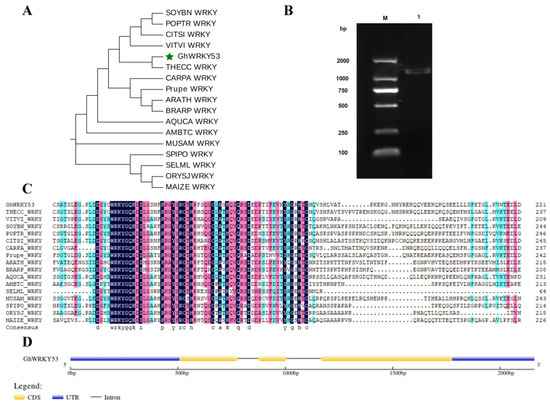
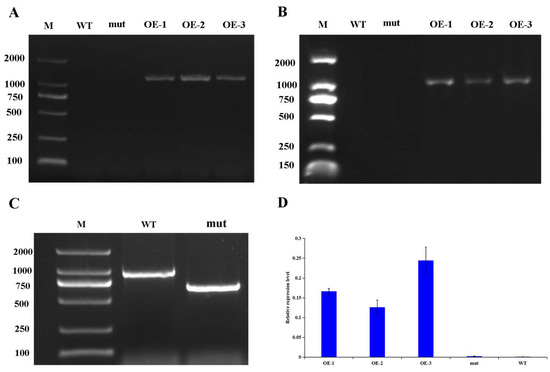
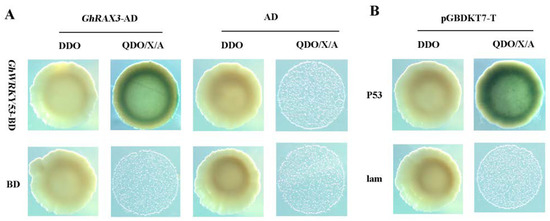
Rapid Alkalization Factor (RALF) is a signaling molecule in plants that plays a crucial role in growth and development, reproductive processes, and responses to both biotic and abiotic stresses. Although RALF peptides have been characterized in Arabidopsis and rice, a comprehensive bioinformatics analysis of the ZmRALF gene family in maize is still lacking. In this study, we identified 20 RALF genes in the maize genome. Sequence alignment revealed significant structural variation among the ZmRALF family genes. Phylogenetic analysis indicates that RALF proteins from Arabidopsis, rice, and maize can be classified into four distinct clades. Duplication events suggest that the expansion of the RALF gene family in maize primarily relies on whole-genome duplication. ZmRALF genes are widely expressed across various tissues; ZmRALF1/15/18/19 are highly expressed in roots, while ZmRALF6/11/14/16 are predominantly expressed in anthers. RNA-seq and RT-qPCR demonstrated that the expression levels of ZmRALF7, ZmRALF9, and ZmRALF13 were significantly up-regulated and down-regulated in response to PEG and NaCl stresses, respectively. Overall, our study provides new insights into the role of the RALF gene family in abiotic stress.
Full article

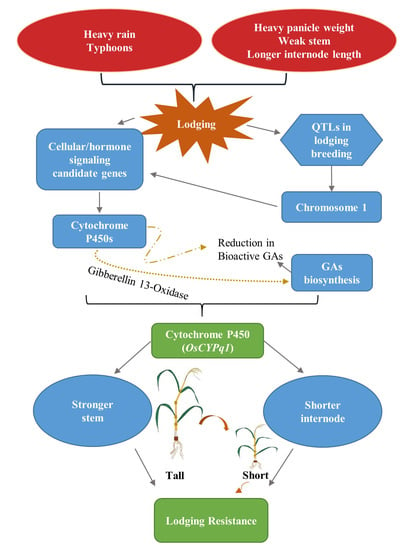
Figure 1




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}