




Biomolecules 2024, 14(5), 528; https://doi.org/10.3390/biom14050528 - 28 Apr 2024
Viewed by 1308
Abstract
►
Show Figures
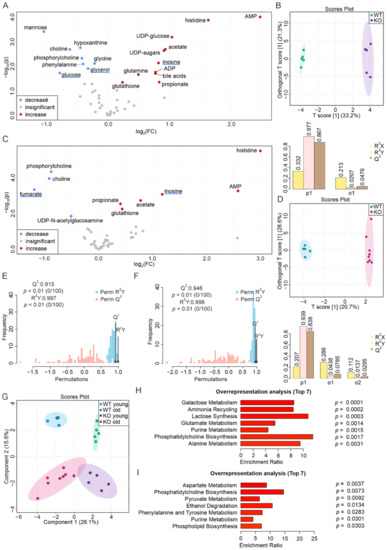
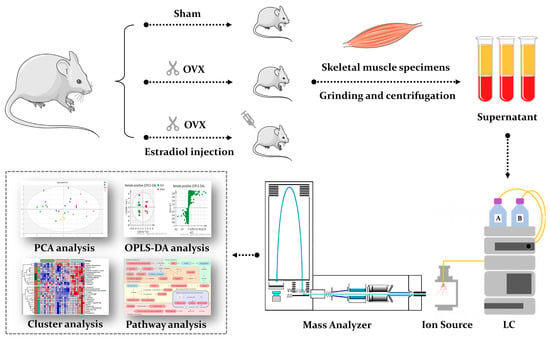
Although both localized nuclear magnetic resonance spectroscopy (MRS) and non-localized nuclear magnetic resonance spectroscopy (NMR) generate the same information, i.e., spectra generated by various groups from the structure of metabolites, they are rarely employed in the same study or by the same research
[...] Read more.
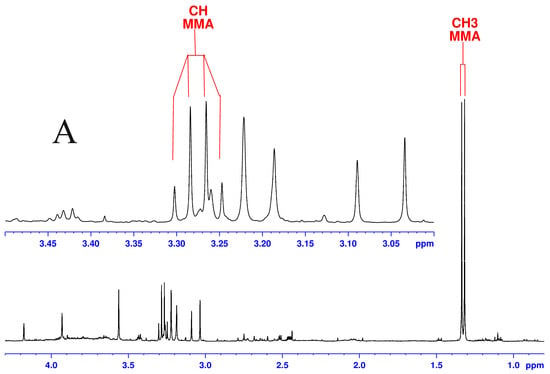
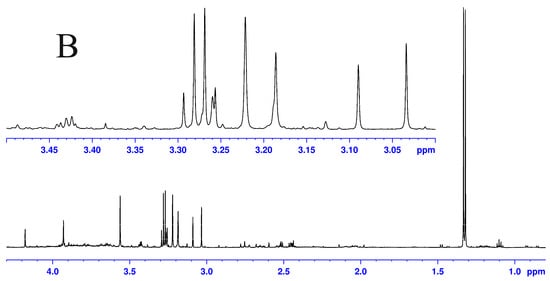
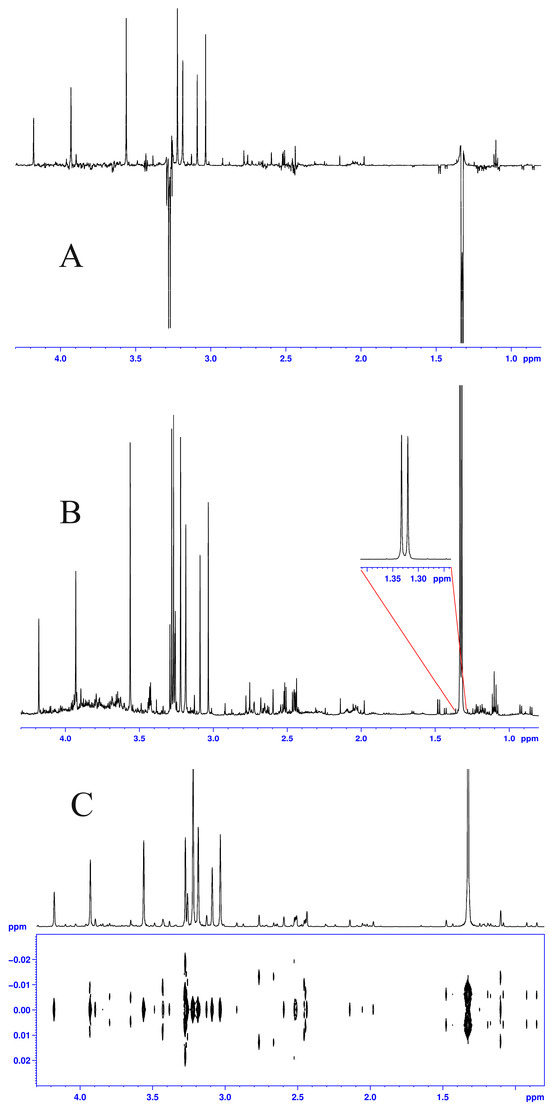
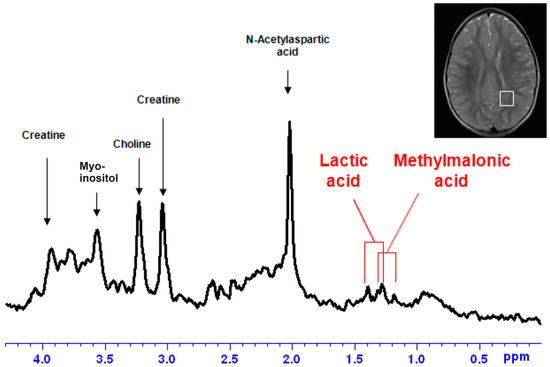
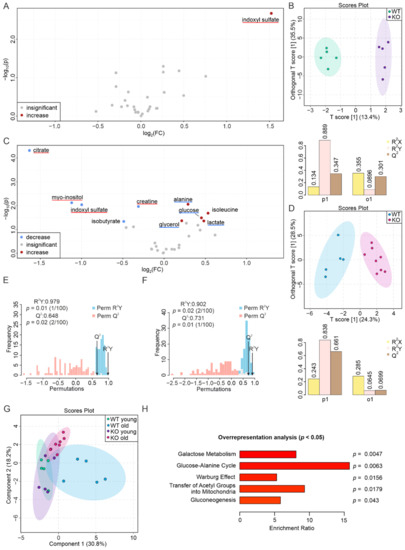
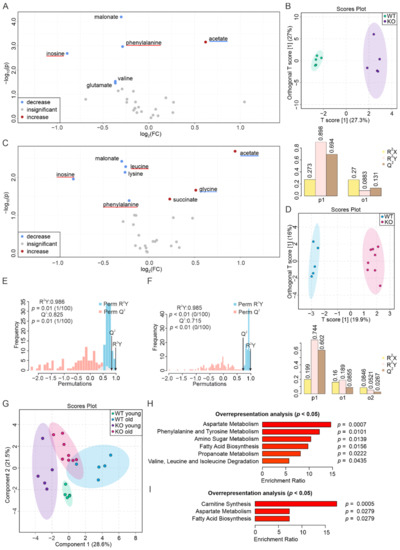
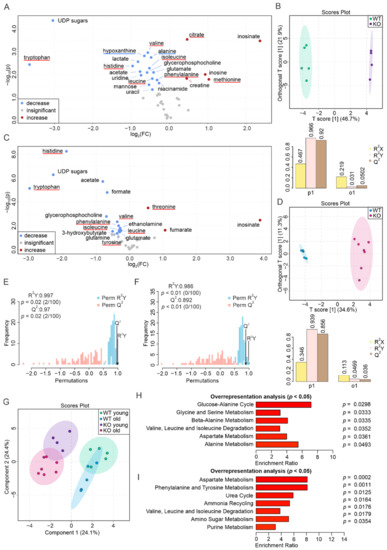
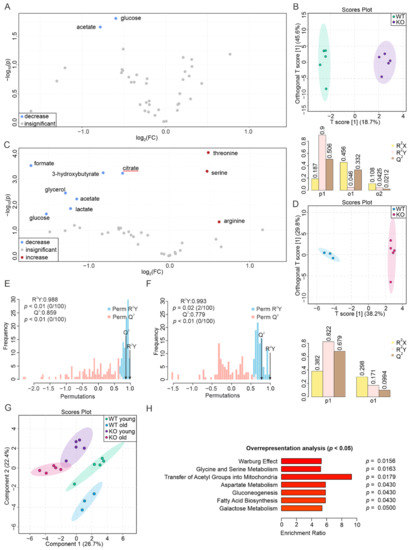
Although both localized nuclear magnetic resonance spectroscopy (MRS) and non-localized nuclear magnetic resonance spectroscopy (NMR) generate the same information, i.e., spectra generated by various groups from the structure of metabolites, they are rarely employed in the same study or by the same research group. As our review reveals, these techniques have never been applied in the same study of methylmalonic acidemia (MMA), propionic acidemia (PA) or vitamin B12 deficiency patients. On the other hand, MRS and NMR provide complementary information which is very valuable in the assessment of the severity of disease and efficiency of its treatment. Thus, MRS provides intracellular metabolic information from localized regions of the brain, while NMR provides extracellular metabolic information from biological fluids like urine, blood or cerebrospinal fluid. This paper presents an up-to-date review of the NMR and MRS studies reported to date for methylmalonic and propionic acidemias. Vitamin B12 deficiency, although in most of its cases not inherited, shares similarities in its metabolic effects with MMA and it is also covered in this review.
Full article
Graphical abstract










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}