





Genes 2024, 15(11), 1462; https://doi.org/10.3390/genes15111462 - 13 Nov 2024
Viewed by 538
Abstract
►
Show Figures
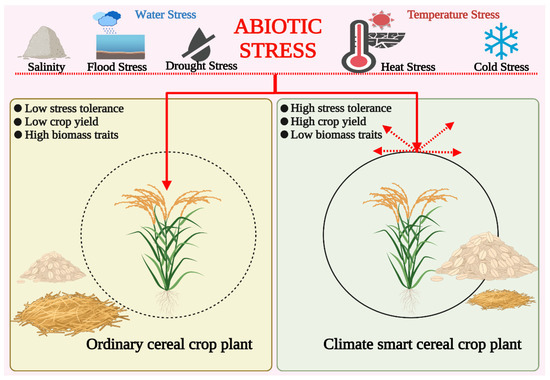
Background/Objectives: Carrot is a major root vegetable in the Apiaceae owing to its abundant carotenoids, antioxidants, vitamins, and minerals. The modern dark orange western carrot was derived from sequential domestication events from the white-rooted wild form to the pale orange-, purple-, or yellow-rooted
[...] Read more.
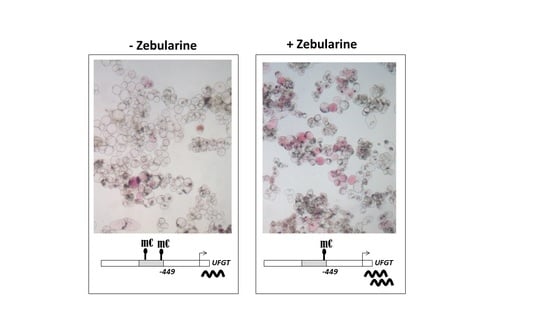
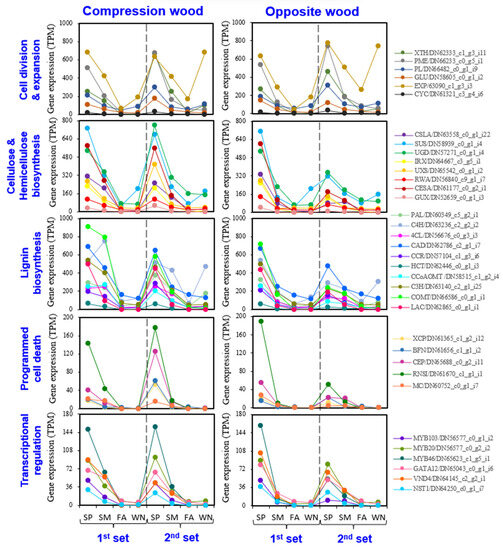
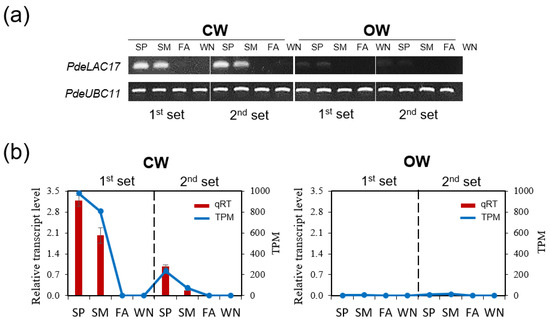
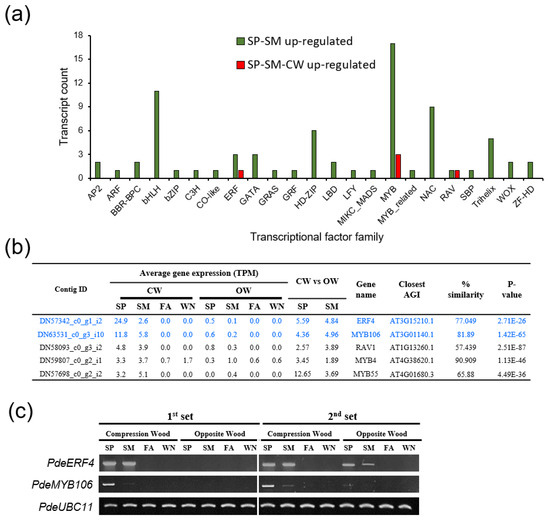
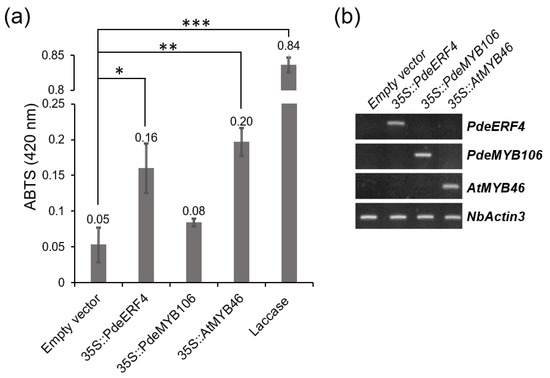
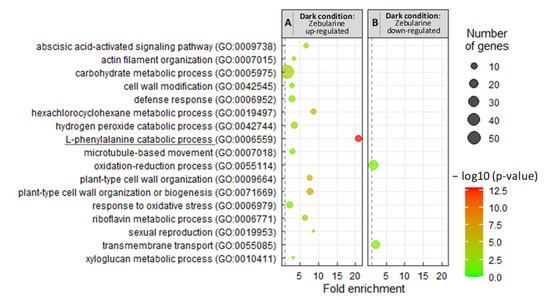
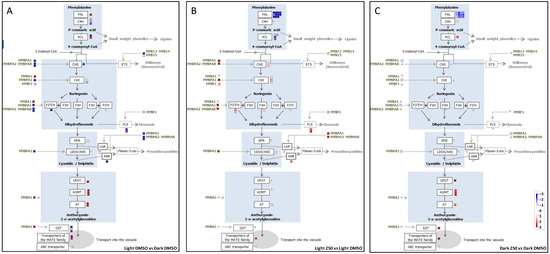
Background/Objectives: Carrot is a major root vegetable in the Apiaceae owing to its abundant carotenoids, antioxidants, vitamins, and minerals. The modern dark orange western carrot was derived from sequential domestication events from the white-rooted wild form to the pale orange-, purple-, or yellow-rooted eastern carrot. Genetic and molecular studies between eastern and western carrots are meager despite their evolutionary relatedness. Methods: Twelve RNA seq libraries obtained from distinct eastern and western cultivars at vegetative and reproductive developmental stages were utilized to identify differentially expressed genes (DEGs) to decode the key molecular genetic changes in carotenoid and flowering pathways. Results: In the carotenoid pathway, an upregulation of the PSY, CRTISO, and LCYE genes was observed in the western cultivar, while the eastern cultivar exhibited a higher abundance of downstream enzymes, particularly CCD and NCED1. These later enzymes are crucial in linking apocarotenoids and xanthin-mediated ABA signaling. In the flowering pathway, we noted a greater expression of DEGs associated with the photoperiod and vernalization pathways in the western cultivar. In contrast, the eastern cultivar displayed a dominance of genes from the autonomous pathway (FLD, LD, FLK, and PEBP) that function to repress FLC. The experimental validation of 12 key genes through quantitative real-time PCR further confirms their functional role in carrots. Conclusions: The identified key regulatory genes in these major pathways are valuable for designing breeding strategies for manipulating carotenoid content and flowering time while developing climate-specific carrots. The knowledge of carotenoid and flowering pathways is advantageous in producing nutritionally improved roots and seeds in carrots across diverse climates.
Full article

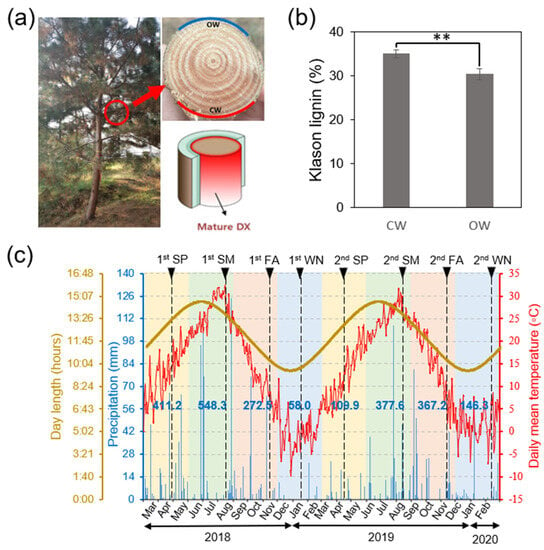
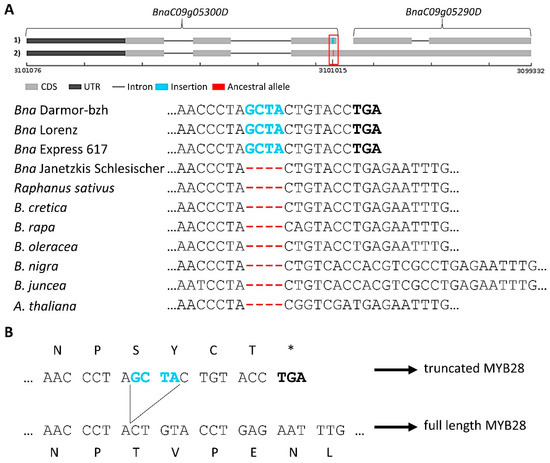
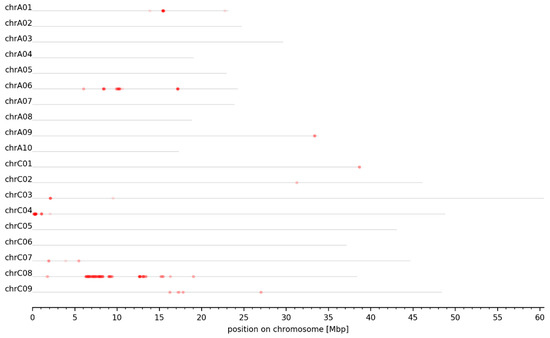
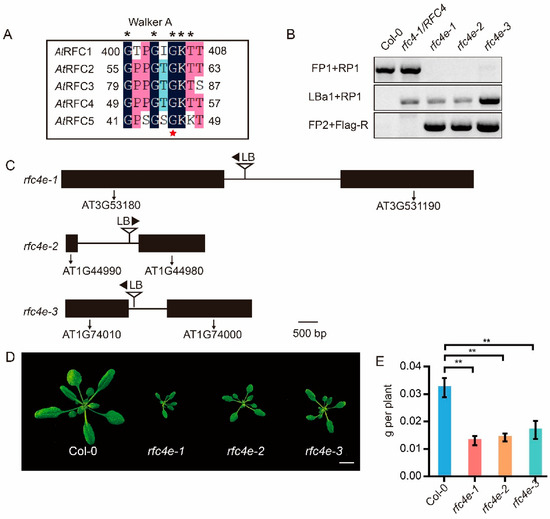
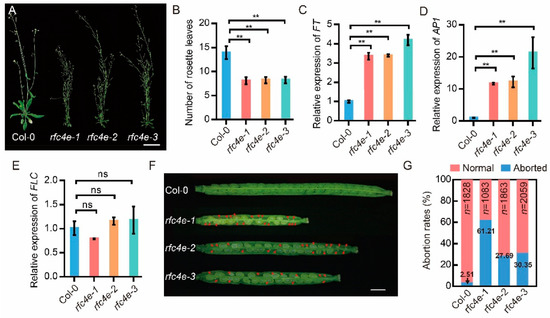
Figure 1
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}