



Animals 2024, 14(16), 2407; https://doi.org/10.3390/ani14162407 - 19 Aug 2024
Viewed by 814
Abstract
►
Show Figures
Africa is home to a wide diversity of locally adapted pig breeds whose genetic architecture offers important insights into livestock adaptation to climate change. However, the majority of these inherent traits have not been fully highlighted. This review presents an overview of the
[...] Read more.
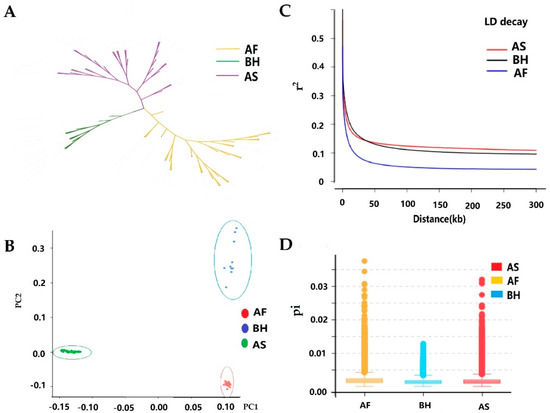
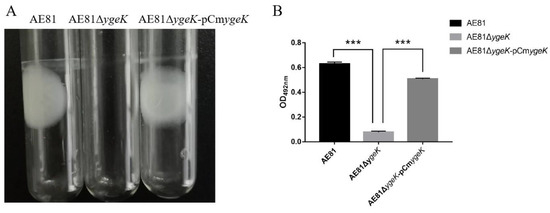
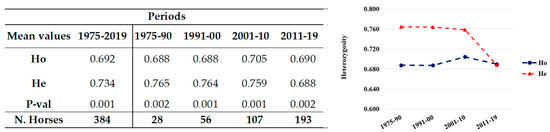
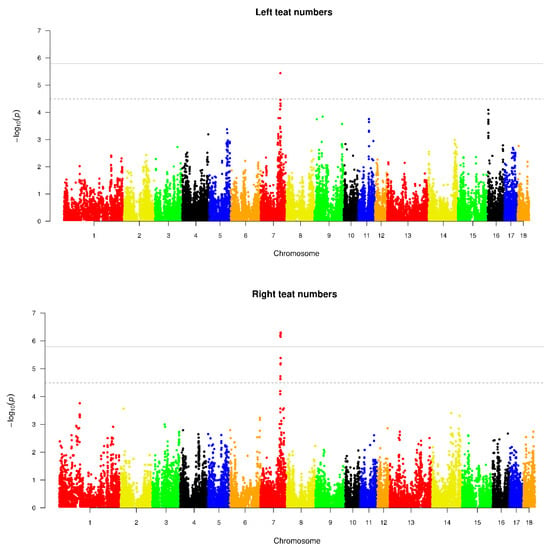
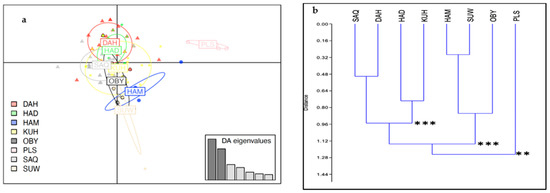
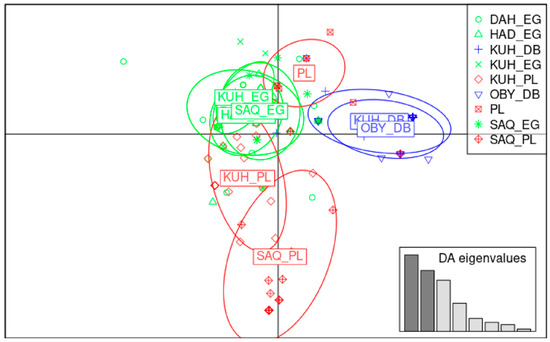
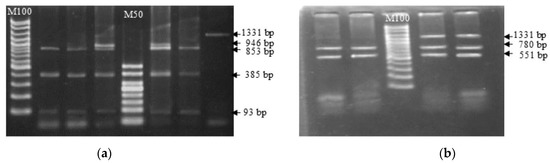
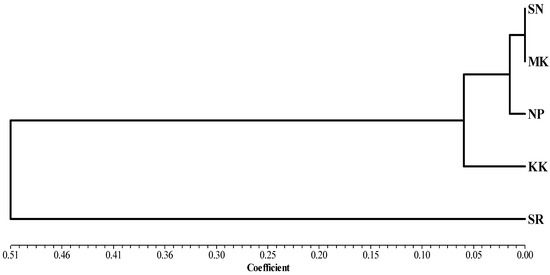
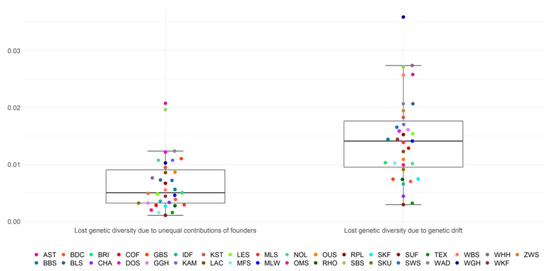
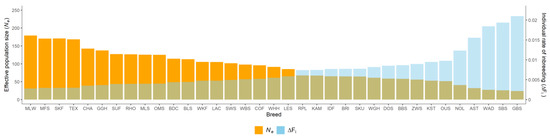
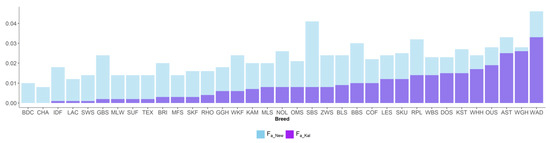
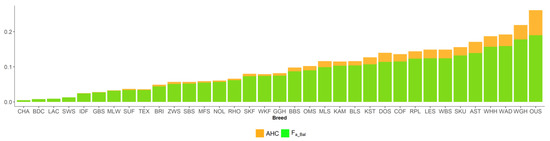
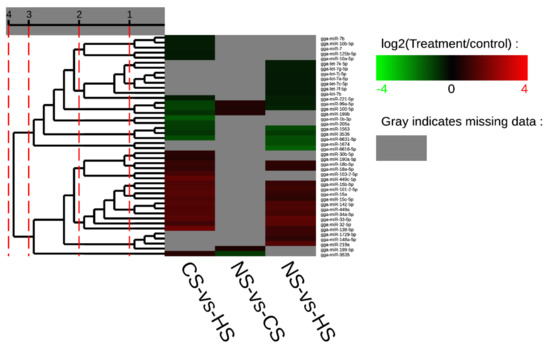
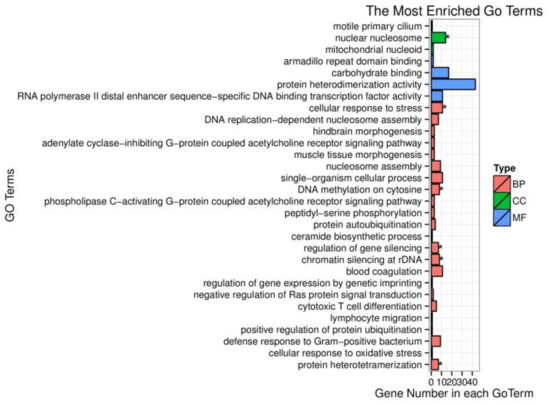
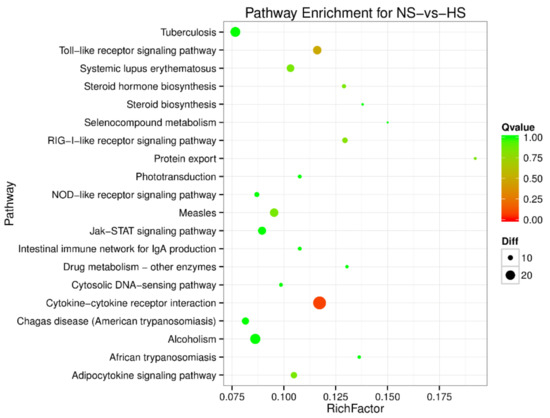
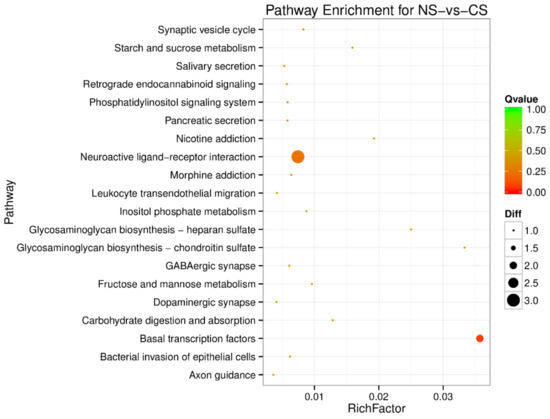
Africa is home to a wide diversity of locally adapted pig breeds whose genetic architecture offers important insights into livestock adaptation to climate change. However, the majority of these inherent traits have not been fully highlighted. This review presents an overview of the current state of African pig genetic resources, providing highlights on their population and production statistics, production system, population diversity indices, and genomic evidence underlying their evolutionary potential. The study results reveal an incomplete characterization of local pig genotypes across the continent. The characterized population, however, demonstrates moderate to high levels of genetic diversity, enough to support breeding and conservation programs. Owing to low genetic differentiation and limited evidence of distinct population structures, it appears that most local pig populations are strains within larger breeds. Genomic evidence has shown a higher number of selection signatures associated with various economically important traits, thus making them potential candidates for climate change adaptation. The reportedly early evidence of hybridization with wild suid groups further suggests untapped insights into disease resistance and resilience traits that need to be illuminated using higher-density markers. Nevertheless, gene introgression from commercial breeds is prevalent across Africa; thus, efforts to realize and utilize these traits must increase before they are permanently depleted.
Full article

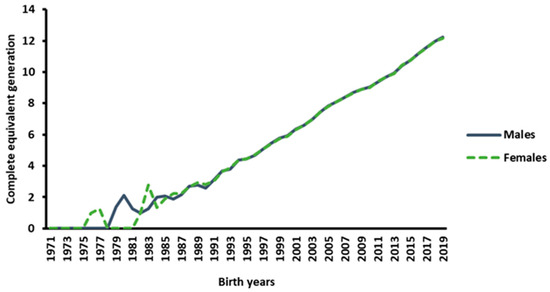
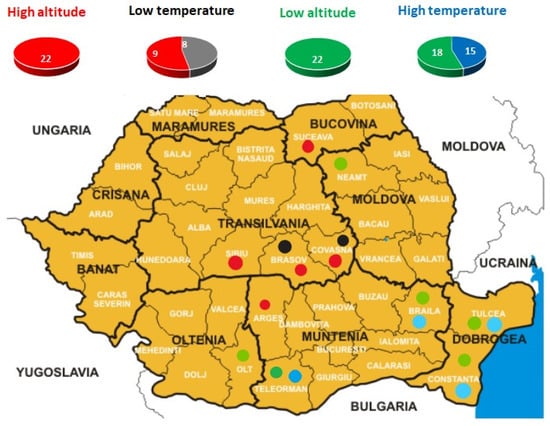
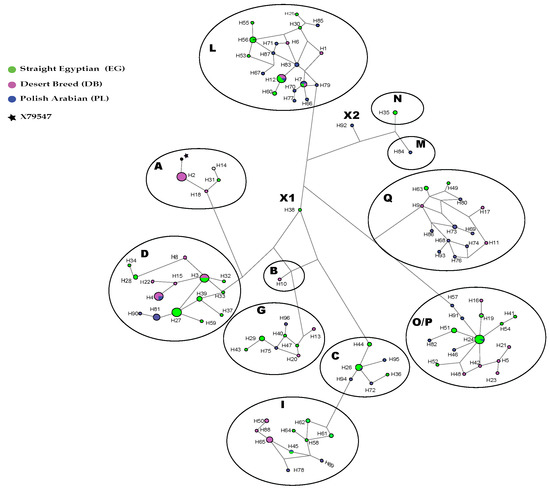
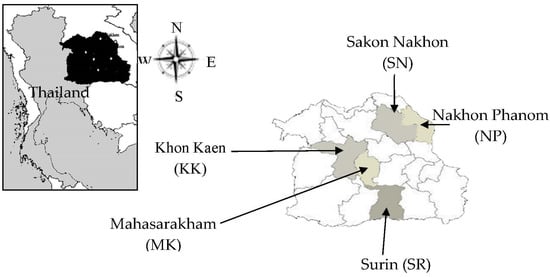
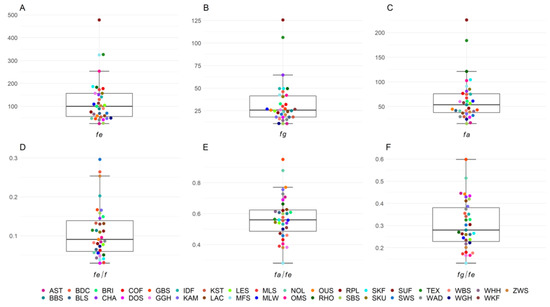
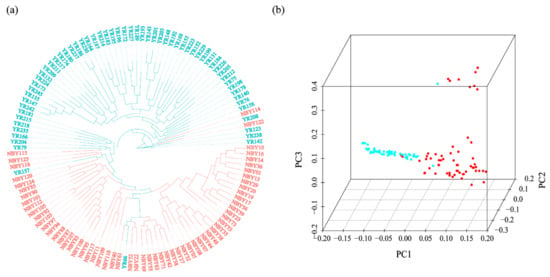
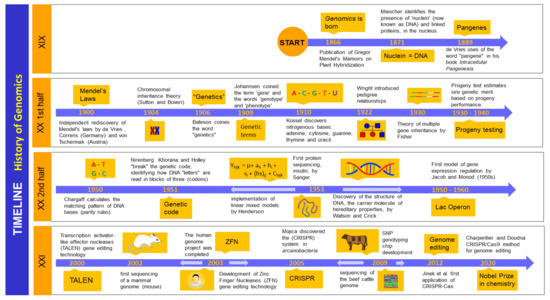
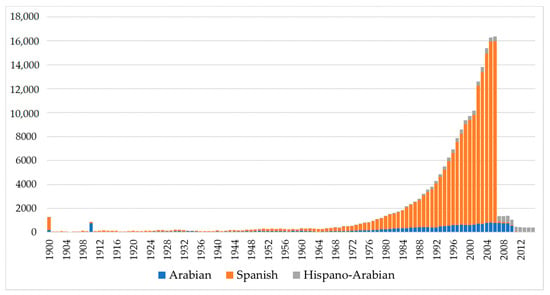
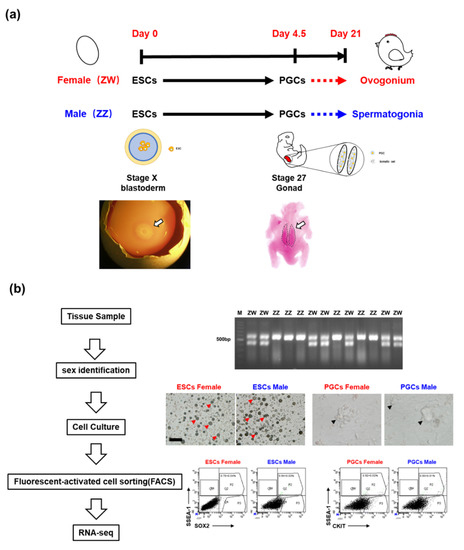
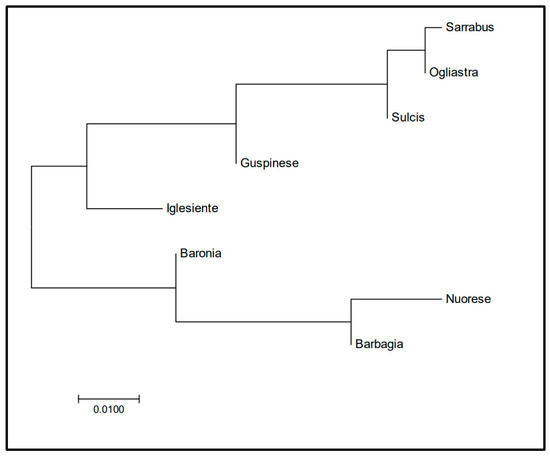
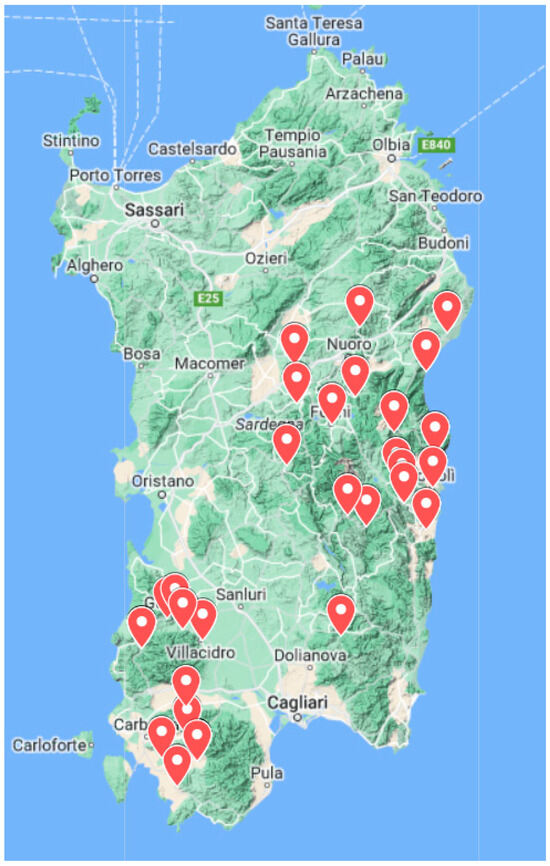
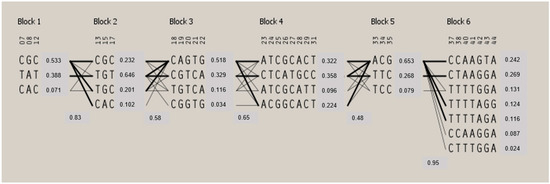
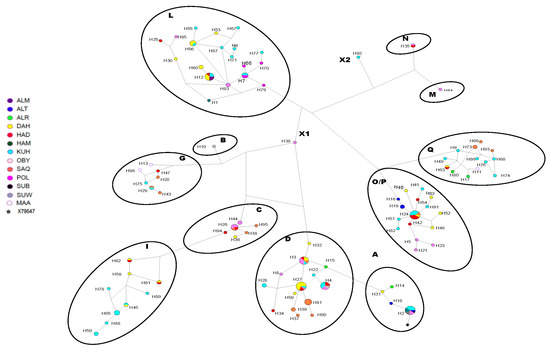
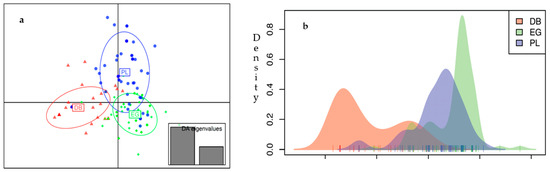
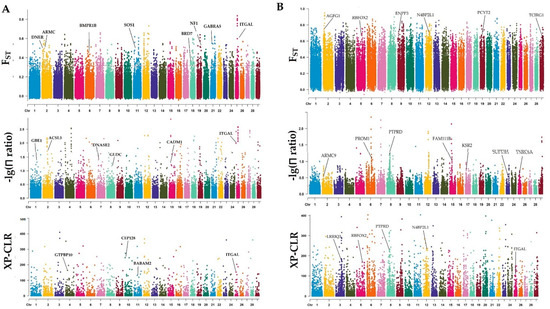
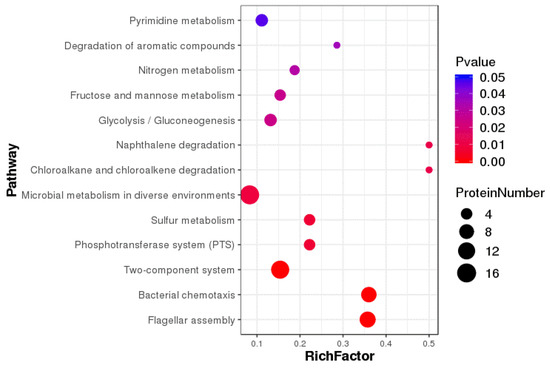
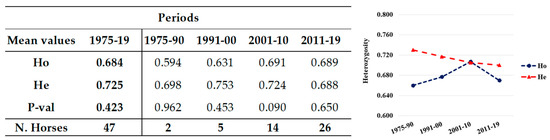
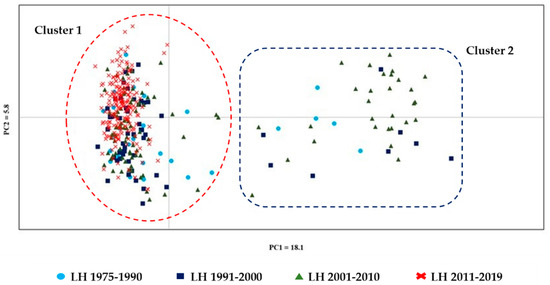
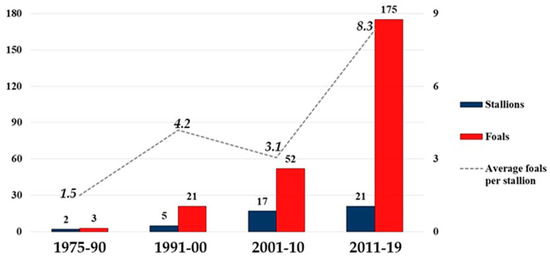
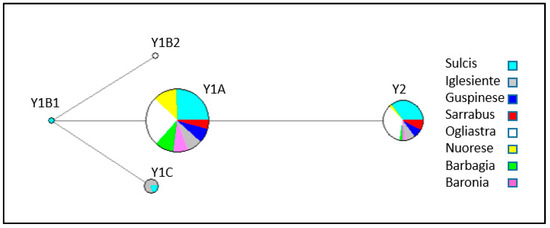
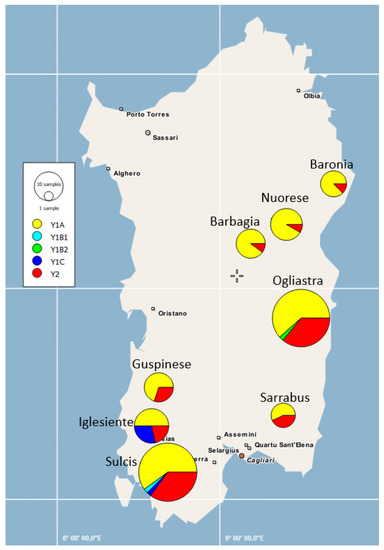
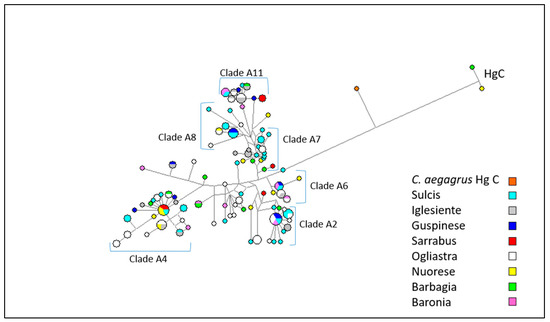
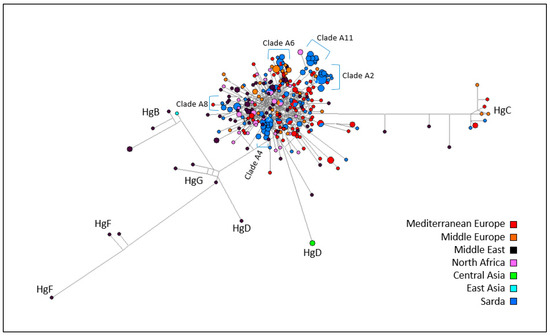
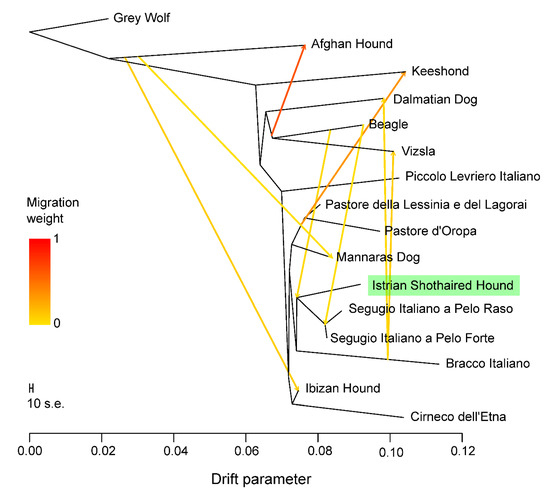
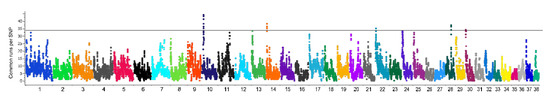
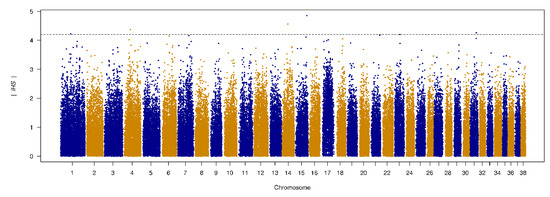
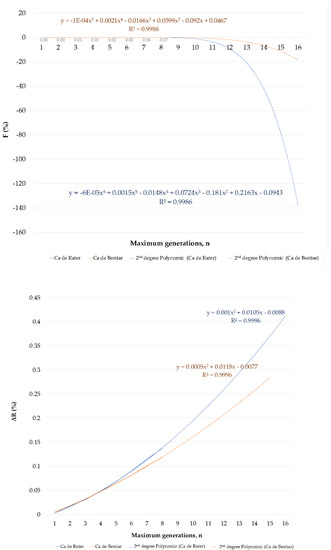
Figure 1






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}